Vyleesi for Women: Clinical Evidence on HSDD
Bremelanotide and Sexual Function
1,247 women studied
Two identical Phase 3 trials (RECONNECT) enrolled 1,247 premenopausal women with hypoactive sexual desire disorder, producing the largest dataset on bremelanotide efficacy.
Kingsberg et al., Obstetrics & Gynecology, 2019
Kingsberg et al., Obstetrics & Gynecology, 2019
If you only read one thing
Vyleesi (bremelanotide) is a self-injectable peptide drug that boosts sexual desire by activating 'wanting' circuits in the brain. It's FDA-approved and actually works — desire went up and distress went down in trials with over 1,200 women. The catch: 4 out of 10 women get nauseous, you have to inject it 45 minutes before sex, and while the improvement is real, it's modest. It won't transform your sex life, but for women with genuine HSDD who haven't found other options helpful, it offers something no other drug does — a peptide that targets desire itself, not just blood flow or serotonin.
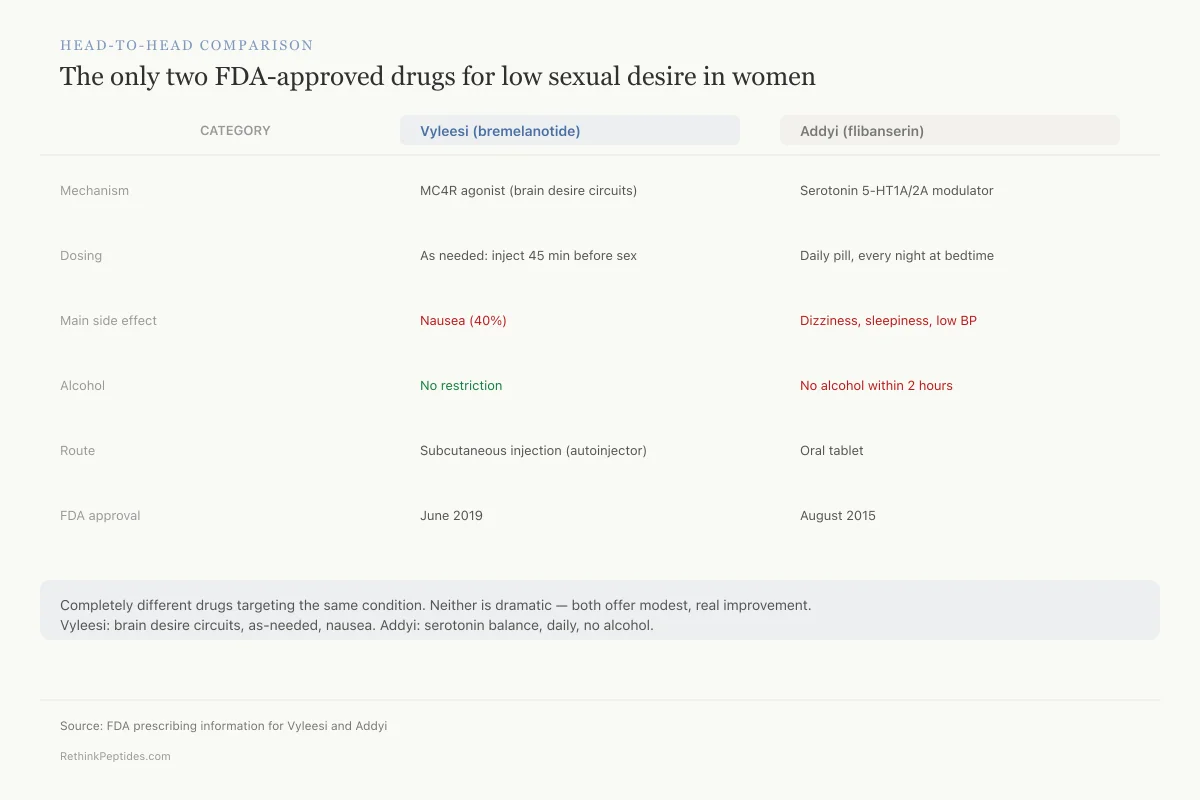
Bremelanotide, sold as Vyleesi, is a synthetic melanocortin receptor agonist and the only injectable FDA-approved treatment for acquired, generalized hypoactive sexual desire disorder (HSDD) in premenopausal women. It received FDA approval in June 2019 based on two Phase 3 RECONNECT trials enrolling 1,247 women.[1] Unlike flibanserin (Addyi), which is taken daily and acts on serotonin receptors, bremelanotide is injected subcutaneously as needed and works through the melanocortin-4 receptor (MC4R) in the central nervous system.
This article is the hub for RethinkPeptides' coverage of bremelanotide and melanocortin-based sexual function research. Each section links to deeper coverage: how bremelanotide works in the CNS, PT-141 as an FDA-approved peptide, PT-141 for male erectile dysfunction, PT-141 side effects, and PT-141 vs PDE5 inhibitors.
Key Takeaways
- Vyleesi came from a tanning drug that accidentally caused erections in its first male volunteers.
- It's the only FDA-approved injection for women whose sexual desire has dropped and causes them distress.
- It works on the brain, not blood flow — it targets the "wanting" circuit itself.
- Trials in over 1,200 women showed real improvements in desire and a real drop in distress.
- About 40% of women feel nauseous after using it — that's the biggest drawback.
- You inject it about 45 minutes before sex, not daily — it's on-demand, not a routine pill.
- Nearly 80% of women who finished the trial volunteered for another year on it.
What is HSDD and why existing treatments fell short
Hypoactive sexual desire disorder is defined as persistently low or absent sexual desire that causes marked personal distress or interpersonal difficulty. It is not simply a low libido; the distress criterion is essential. Epidemiological estimates suggest HSDD affects 8-14% of premenopausal women when the distress criterion is applied, making it the most common female sexual complaint reported in clinical settings.[2]
Before bremelanotide, the only FDA-approved pharmacotherapy for premenopausal HSDD was flibanserin (Addyi, approved 2015), a daily oral medication that modulates serotonin 5-HT1A and 5-HT2A receptors. Flibanserin's efficacy was modest (about 0.5 additional satisfying sexual events per month versus placebo), its alcohol restriction was burdensome, and its mechanism had no clear relationship to the neurobiology of sexual desire.[2]
The melanocortin pathway offered a fundamentally different approach. Melanocortin receptors, particularly MC4R, are expressed in brain regions directly involved in sexual motivation and arousal, including the medial preoptic area, ventromedial hypothalamus, and paraventricular nucleus. A drug targeting this system would, in theory, act on circuits that process desire itself rather than modulating neurotransmitters with indirect effects.
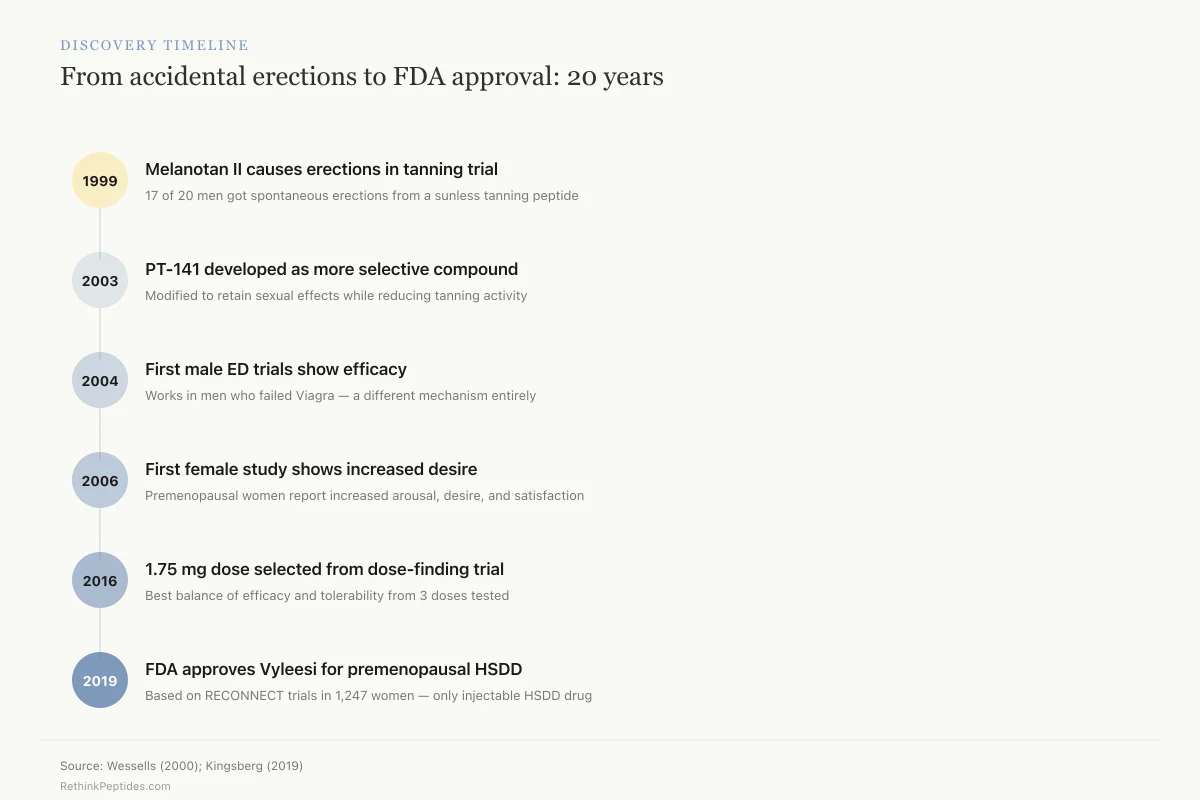
From tanning peptide to sexual function: the accidental discovery
Bremelanotide's origin story is one of the more unusual in pharmaceutical history. In the late 1990s, researchers at the University of Arizona were studying Melanotan II, a synthetic analog of alpha-melanocyte-stimulating hormone (alpha-MSH) designed to produce a sunless tan by stimulating melanocortin-1 receptors (MC1R) in skin.
During a 1999 clinical study, Wessells et al. documented that Melanotan II caused spontaneous penile erections in male volunteers, an effect unrelated to its intended tanning purpose. The study found that subcutaneous Melanotan II produced erections in 17 of 20 subjects, along with increased sexual desire and motivation.[3] This was a striking departure from existing erectile dysfunction treatments like sildenafil (Viagra), which increase blood flow to the penis but do not affect desire. Melanotan II appeared to activate central nervous system pathways involved in wanting, not just in the mechanics of arousal.
Hadley documented in 2005 how this serendipitous finding redirected an entire research program, establishing that melanocortin receptor agonists regulate sexual functions in both men and women through central mechanisms distinct from peripheral vasodilators.[4] For the full story of Melanotan II's unexpected effects, see Melanotan II and Erections: The Accidental Discovery.
Discovery Timeline
From accidental erections to FDA approval: 20 years
Melanotan II causes erections in tanning trial
17 of 20 men got spontaneous erections from a sunless tanning peptide
PT-141 developed as more selective compound
Modified to retain sexual effects while reducing tanning activity
First male ED trials show efficacy
Works in men who failed Viagra — a different mechanism entirely
First female study shows increased desire
Premenopausal women report increased arousal, desire, and satisfaction
1.75 mg dose selected from dose-finding trial
Best balance of efficacy and tolerability from 3 doses tested
FDA approves Vyleesi for premenopausal HSDD
Based on RECONNECT trials in 1,247 women — only injectable HSDD drug
Source: Wessells (2000); Kingsberg (2019)
 View as image
View as imagePT-141 becomes bremelanotide: the preclinical evidence
Melanotan II was a non-selective melanocortin agonist with tanning, appetite, and sexual effects. Researchers needed a compound that was more selective for sexual function. PT-141 (later named bremelanotide) was developed as a cyclic heptapeptide metabolite of Melanotan II with reduced melanocortin-1 activity (less tanning) but retained MC4R activity (sexual function).
Molinoff et al. described PT-141 in 2003 as a melanocortin agonist for the treatment of sexual dysfunction, establishing its receptor binding profile and initial pharmacology.[5] The preclinical work was striking. Pfaus et al. showed in 2004 that a melanocortin receptor agonist selectively facilitated sexual solicitation behavior in female rats without affecting locomotion or anxiety, suggesting the sexual effects were specific rather than a byproduct of general arousal.[6]
Pfaus expanded on this in 2007 with a comprehensive review of bremelanotide's preclinical CNS effects on female sexual function, demonstrating that the drug increased lordosis (receptive posture) and solicitation in female rats by acting on hypothalamic melanocortin circuits, with effects blocked by MC4R antagonists.[7] Shadiack et al. reviewed the broader melanocortin approach in 2007, noting that bremelanotide's mechanism of action was fundamentally different from all existing sexual dysfunction treatments because it targeted the central motivational circuitry rather than peripheral physiology.[8]
Early human trials: male erectile dysfunction data
Before pivoting to female HSDD, bremelanotide was studied in male erectile dysfunction. Diamond et al. conducted a double-blind, placebo-controlled evaluation in 2004 of intranasal PT-141 in healthy males and men with mild-to-moderate ED, finding dose-dependent increases in erection rigidity and duration.[9]
Rosen et al. published a parallel study in 2004 evaluating subcutaneous PT-141 in men with inadequate response to sildenafil, finding that PT-141 produced erections in men for whom Viagra had failed.[10] Diamond et al. then showed in 2005 that co-administration of low-dose intranasal PT-141 with sildenafil enhanced the erectile response beyond either agent alone, suggesting complementary mechanisms.[11]
Safarinejad's 2008 randomized trial confirmed this in a larger male cohort: bremelanotide salvaged erectile function in men who had failed sildenafil, with statistically significant improvements in erection quality and sexual satisfaction.[12] For a full review of the male data, see PT-141 for Male Erectile Dysfunction: Research Beyond Its Approved Use.
However, intranasal bremelanotide raised blood pressure in some subjects. This safety signal led to reformulation as a subcutaneous injection, which had a more favorable hemodynamic profile, and a strategic pivot toward female HSDD where the unmet need was greater.
The pivot to female HSDD
Diamond et al. published the first female study in 2006, showing that bremelanotide had a significant effect on subjective sexual response in premenopausal women with sexual arousal disorder. Women receiving bremelanotide reported increased genital arousal, desire, and overall sexual satisfaction compared to placebo.[13]
Clayton et al. conducted the pivotal dose-finding trial in 2016, a randomized, placebo-controlled study testing 0.75 mg, 1.25 mg, and 1.75 mg doses in premenopausal women with HSDD or female sexual arousal disorder. The 1.75 mg dose showed the strongest efficacy signal with acceptable tolerability, and was selected for Phase 3 development.[14]
The RECONNECT Phase 3 trials: what 1,247 women showed
The pivotal RECONNECT program consisted of two identical Phase 3, randomized, double-blind, placebo-controlled, multicenter trials in premenopausal women with acquired, generalized HSDD of at least 6 months' duration.
Study design
- 1,247 premenopausal women randomized (roughly 2:1 bremelanotide 1.75 mg to placebo)
- Self-administered subcutaneous injection, as needed, at least 45 minutes before anticipated sexual activity
- Maximum one dose per 24 hours, maximum 8 doses per month
- 24-week treatment period
- Age range 19-56 years (mean age 39), 86% White, 12% Black
- Co-primary endpoints: change in Female Sexual Function Index desire domain (FSFI-D) and Female Sexual Distress Scale desire/arousal/orgasm item (FSDS-DAO Item 13)
Efficacy results
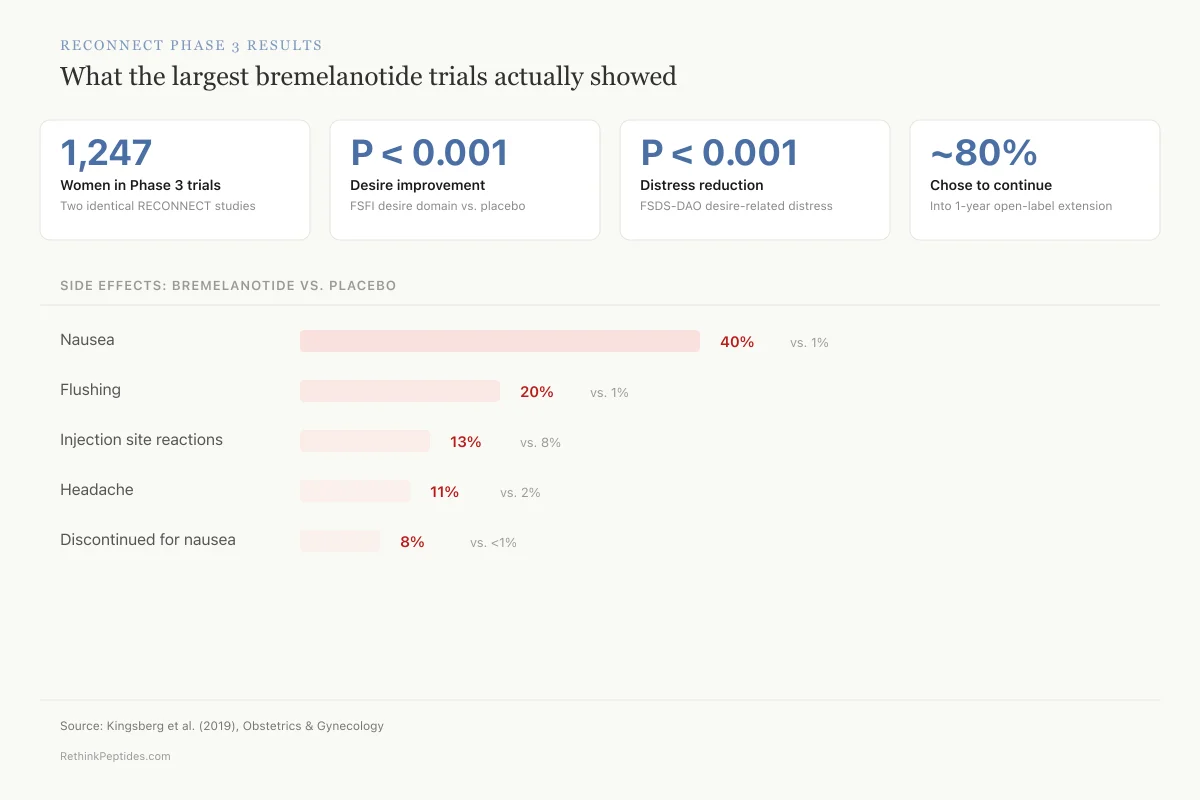
Both trials met both co-primary endpoints. Kingsberg et al. reported in 2019 that bremelanotide-treated women showed statistically significant increases in sexual desire (FSFI-D score improvement, integrated studies P < 0.001) and statistically significant reductions in distress related to low sexual desire (FSDS-DAO Item 13, integrated studies P < 0.001).[1]
Simon et al. published prespecified and integrated subgroup analyses in 2022, finding consistent efficacy across age groups, BMI categories, race, duration of HSDD, and baseline severity. The treatment effect was not driven by any single demographic subgroup.[15]
RECONNECT Phase 3 Results
What the largest bremelanotide trials actually showed
1,247
Women in Phase 3 trials
Two identical RECONNECT studies, randomized 2:1
P < 0.001
Desire score improvement
FSFI desire domain, bremelanotide vs. placebo
P < 0.001
Distress reduction
FSDS-DAO Item 13 (desire-related distress)
~80%
Chose to continue
Volunteered for 1-year open-label extension
Side effects: bremelanotide vs. placebo
Source: Kingsberg et al. (2019), Obstetrics & Gynecology
 View as image
View as imageSafety profile
The safety data showed a clear pattern. The most common adverse effect was nausea, occurring in 40% of bremelanotide-treated women versus 1% on placebo. Other common adverse effects included flushing (20% vs 1%), injection site reactions (13% vs 8%), and headache (11% vs 2%).[1]
Safety
Moderate4 in 10 women experience nausea
Concern
Nausea is the most common side effect of bremelanotide, affecting 40% of women (vs. 1% on placebo). This is an on-target effect — the same melanocortin-4 receptor that controls desire also controls nausea signaling in the brainstem. You can't fully separate the two.
What the research says
Most nausea episodes are mild-to-moderate and decrease with repeated dosing. However, 8% of women discontinued the trial due to nausea alone. Anti-nausea medication before injection may help some women, though this wasn't formally tested.
Particularly relevant for: All women considering bremelanotide
What to do
Expect nausea with the first few doses. If it doesn't improve after several uses, the drug may not be tolerable for you. The overall discontinuation rate for adverse events was 18% — about 1 in 5 women stopped because of side effects.
Kingsberg et al. (2019), Obstetrics & Gynecology
The 40% nausea rate is high. However, most episodes were mild to moderate and tended to decrease with repeated dosing. Still, 8% of bremelanotide-treated women discontinued the study due to nausea alone, and the overall discontinuation rate for adverse reactions was 18% (versus 2% for placebo). For a full analysis of tolerability, see PT-141 Side Effects: Nausea, Blood Pressure, and Skin Darkening.
Transient blood pressure increases of 2-3 mmHg systolic were observed. Bremelanotide carries a labeling restriction for women with uncontrolled hypertension or known cardiovascular disease. For more context on melanocortin receptor activation and blood pressure, see Melanocortin Peptides and Blood Pressure: The Hypertensive Risk.
What the numbers mean in practice
The efficacy signal was statistically significant but clinically modest. The improvement in desire scores, while real, did not transform the sexual experience for most participants. This is consistent with the complexity of sexual desire, which involves psychological, relational, hormonal, and neurological factors that no single peptide can fully address.
Cipriani et al. provided an independent evaluation in 2023, concluding that bremelanotide offers a genuine but limited improvement in desire and distress for women with HSDD, positioned as one tool among many rather than a standalone solution.[16]
Long-term data: the open-label extension
Koochaki et al. reported in 2021 on the RECONNECT exit study, documenting patient experience among women who completed the Phase 3 program. Nearly 80% of women who finished the controlled study volunteered for an additional 52-week open-label extension, suggesting that the drug's perceived benefit outweighed the nausea burden for most completers.[17]
Self-reported satisfaction was moderate. Women reported improvements in desire and arousal that were meaningful to them personally, even when the magnitude of change on validated scales was statistically modest. The exit study data highlights a gap between clinical trial endpoints (which measure average group change on standardized questionnaires) and individual patient experience.
The nausea rate decreased over time in the extension study, consistent with a tolerance effect. However, a subset of women continued to experience nausea throughout the treatment period, and the cumulative dropout rate over 18+ months of use has not been publicly reported in detail.
How bremelanotide works: the MC4R mechanism
Bremelanotide is a cyclic heptapeptide analog of alpha-MSH that acts as an agonist at melanocortin-3 and melanocortin-4 receptors (MC3R and MC4R). Its effects on sexual function are primarily mediated through MC4R in the central nervous system.
Pfaus et al. published a neurobiology review in 2022 detailing how bremelanotide modulates brain circuits involved in sexual desire. Using fMRI in women with HSDD, researchers showed that bremelanotide altered activation patterns in brain regions associated with desire and arousal, including the prefrontal cortex, insula, and anterior cingulate cortex.[18]
This is fundamentally different from how PDE5 inhibitors (sildenafil, tadalafil) work. PDE5 inhibitors increase blood flow to genital tissue by inhibiting the enzyme that breaks down cyclic GMP. They do not cross the blood-brain barrier at therapeutic doses and do not affect desire. Bremelanotide crosses the blood-brain barrier, activates MC4R on neurons in desire-related circuits, and produces subjective wanting. For a direct mechanism comparison, see PT-141 vs PDE5 Inhibitors (Viagra): Fundamentally Different Mechanisms.
The MC4R mechanism also explains bremelanotide's side effects. MC4R is expressed throughout the hypothalamus and brainstem, including in nuclei controlling nausea (area postrema), blood pressure (sympathetic outflow), and energy balance. Activating MC4R broadly produces the desired sexual motivation effect but also triggers nausea, flushing, and transient hypertension as on-target side effects.
Head-to-Head Comparison
The only two FDA-approved drugs for low sexual desire in women
Vyleesi
Addyi
Melanocortin-4 receptor agonist (brain desire circuits)
Serotonin 5-HT1A/5-HT2A modulator (neurotransmitter balance)
As needed: inject 45 min before sex
Daily pill, every night at bedtime
Nausea (40%)
Dizziness, sleepiness, low BP
None
No alcohol within 2 hours
Subcutaneous injection (autoinjector)
Oral tablet
June 2019
August 2015
Premenopausal women with HSDD
Premenopausal women with HSDD
Bottom line: Completely different drugs targeting the same condition. Vyleesi works on brain desire circuits and is used as-needed but causes nausea in 4 out of 10 women. Addyi works on serotonin balance and is taken daily but restricts alcohol. Neither is a dramatic solution — both offer modest, real improvement.
Source: FDA prescribing information for Vyleesi and Addyi
 View as image
View as imageLimitations of the evidence
Several limitations shape how the bremelanotide evidence should be interpreted:
Population studied. The RECONNECT trials enrolled only premenopausal women with acquired, generalized HSDD. Bremelanotide is not FDA-approved for postmenopausal women, men, or women whose low desire is lifelong rather than acquired. Separate data exists for male ED (see above), but the female indication remains narrow.
Nausea as a confounder. The 40% nausea rate in the active arm raises the possibility of functional unblinding. If women could tell they were on the active drug because of nausea, their self-reported desire scores might be influenced by expectation effects. The trials did not include an active placebo control that would produce nausea without sexual effects.
Clinical versus statistical significance. The improvement in FSFI-D scores was statistically significant but numerically small. Whether a 0.3-0.5 point improvement on a 6-point desire subscale translates to meaningful real-world change for individual women is debated.
Limited diversity. The study population was 86% White. The Phase 3 subgroup analyses by Simon et al. showed consistent effects across racial groups, but the sample sizes for non-White subgroups were small.
On-demand model. Bremelanotide requires self-injection 45 minutes before sexual activity. This on-demand model assumes predictability in sexual encounters that may not reflect how desire works for many women, particularly those whose desire is responsive rather than spontaneous.
The melanocortin system beyond sexual function
Bremelanotide's target, the melanocortin system, extends well beyond sexual function. MC4R is a central regulator of energy homeostasis: loss-of-function MC4R mutations are the most common monogenic cause of human obesity. Alpha-MSH, the endogenous ligand from which bremelanotide derives, also has anti-inflammatory and immunomodulatory properties. This broad biology explains both bremelanotide's therapeutic effects and its side effects.
The connection between melanocortin signaling and appetite is relevant. MC4R activation suppresses feeding, which is why bremelanotide causes nausea and mild weight loss in some users. Spana et al. published data in 2022 from two Phase 1 trials showing a modest body weight reduction in obese women treated with bremelanotide, though the drug is not being developed for weight management. The overlap between melanocortin-regulated desire, appetite, and energy balance reflects the evolutionary integration of reproductive and metabolic systems. Animals do not seek mates when starving; the melanocortin system enforces this priority hierarchy.
For more on the melanocortin system's metabolic roles, see How Bremelanotide Works: A Central Nervous System Approach to Desire.
Where bremelanotide fits in treatment
Bremelanotide occupies a specific niche: it is the only available treatment for premenopausal HSDD that acts on central motivational circuits and is used as-needed rather than daily. For women who have tried flibanserin without adequate response, or who prefer not to take a daily medication, bremelanotide offers a mechanistically distinct alternative.
The drug works through a peptide-receptor system (melanocortin) that is biologically involved in sexual motivation. This is not a repurposed antidepressant or an accidental finding from another drug class. Bremelanotide was developed specifically because melanocortin receptor activation modulates desire circuitry in the brain, and the clinical data confirms that this mechanism produces measurable effects on desire and distress, albeit with a significant nausea burden.
The broader question is whether the melanocortin approach can be refined. Next-generation melanocortin agonists with greater MC4R selectivity or biased agonism (activating desire-related signaling while minimizing nausea-related signaling) could improve the therapeutic window. For now, bremelanotide remains the proof-of-concept that central peptide systems can be pharmacologically targeted to modulate human sexual desire.
For broader context on related peptide approaches to sexual function, see Oxytocin and Male Sexual Function: The Overlooked Connection.
The Bottom Line
Bremelanotide (Vyleesi) is a synthetic melanocortin receptor agonist that was FDA-approved in 2019 for premenopausal HSDD based on two Phase 3 trials in 1,247 women. It produces statistically significant improvements in sexual desire and reductions in desire-related distress. The 40% nausea rate is a significant tolerability limitation. The drug originated from the accidental discovery that Melanotan II tanning peptides caused erections in men, leading to two decades of research on melanocortin receptors in sexual function. Its mechanism of action, working through MC4R in brain desire circuits, is fundamentally different from PDE5 inhibitors or serotonin-targeting drugs.
Sources & References
- 1RPEP-04281·Kingsberg, Sheryl A et al. (2019). “Bremelanotide for the Treatment of Hypoactive Sexual Desire Disorder: Two Randomized Phase 3 Trials..” Obstetrics and gynecology.Study breakdown →PubMed →↩
- 2RPEP-02688·Kingsberg, Sheryl A et al. (2015). “The Female Sexual Response: Current Models, Neurobiological Underpinnings and Agents Currently Approved or Under Investigation for the Treatment of Hypoactive Sexual Desire Disorder..” CNS drugs.Study breakdown →PubMed →↩
- 3RPEP-00633·Wessells, H et al. (2000). “Melanocortin receptor agonists, penile erection, and sexual motivation: human studies with Melanotan II..” International journal of impotence research.Study breakdown →PubMed →↩
- 4RPEP-01041·Hadley, Mac E (2005). “The Accidental Discovery That a Tanning Peptide Enhances Sexual Function in Both Sexes.” Peptides.Study breakdown →PubMed →↩
- 5RPEP-00849·Molinoff, P B et al. (2003). “PT-141: A Melanocortin Peptide That Treats Sexual Dysfunction Through the Brain.” Annals of the New York Academy of Sciences.Study breakdown →PubMed →↩
- 6RPEP-00959·Pfaus, James G et al. (2004). “PT-141 Melanocortin Agonist Selectively Enhances Female Sexual Desire in Rats.” Proceedings of the National Academy of Sciences of the United States of America.Study breakdown →PubMed →↩
- 7RPEP-01279·Pfaus, James et al. (2007). “Bremelanotide (PT-141) for Female Sexual Dysfunction: Comprehensive Preclinical CNS Mechanism Review.” The journal of sexual medicine.Study breakdown →PubMed →↩
- 8RPEP-01288·Shadiack, Annette M et al. (2007). “Melanocortins for Both Male AND Female Sexual Dysfunction: PT-141 and Beyond.” Current topics in medicinal chemistry.Study breakdown →PubMed →↩
- 9RPEP-00877·Unknown (2004). “Diamond 2004 Doubleblind Placebocontrolled Evaluation Off.” .Study breakdown →↩
- 10RPEP-00969·Rosen, R C et al. (2004). “PT-141 Subcutaneous Injection Safely Induces Erection in Erectile Dysfunction Patients.” International journal of impotence research.Study breakdown →PubMed →↩
- 11RPEP-01028·Diamond, L E et al. (2005). “Combining Low-Dose PT-141 Nasal Spray With Viagra: Synergistic Erectile Effect.” Urology.Study breakdown →PubMed →↩
- 12RPEP-01413·Safarinejad, Mohammad Reza et al. (2008). “Bremelanotide Rescues Men Who Fail Viagra: A Brain Approach When Blood Flow Drugs Don't Work.” The Journal of urology.Study breakdown →PubMed →↩
- 13RPEP-01128·Diamond, Lisa E et al. (2006). “PT-141 (Bremelanotide) Improves Sexual Arousal in Women With Sexual Arousal Disorder.” The journal of sexual medicine.Study breakdown →PubMed →↩
- 14RPEP-02899·Clayton, Anita H et al. (2016). “Bremelanotide for female sexual dysfunctions in premenopausal women: a randomized, placebo-controlled dose-finding trial..” Women's health (London.Study breakdown →PubMed →↩
- 15RPEP-06503·Simon, James A et al. (2022). “Prespecified and Integrated Subgroup Analyses from the RECONNECT Phase 3 Studies of Bremelanotide..” Journal of women's health (2002).Study breakdown →PubMed →↩
- 16RPEP-06806·Cipriani, Sarah et al. (2023). “An evaluation of bremelanotide injection for the treatment of hypoactive sexual desire disorder..” Expert opinion on pharmacotherapy.Study breakdown →PubMed →↩
- 17RPEP-05507·Koochaki, Patricia et al. (2021). “Women's Experience With Bremelanotide for Low Sexual Desire: Increased Desire and Physical Arousal.” Journal of women's health (2002).Study breakdown →PubMed →↩
- 18RPEP-06431·Pfaus, James G et al. (2022). “The neurobiology of bremelanotide for the treatment of hypoactive sexual desire disorder in premenopausal women..” CNS spectrums.Study breakdown →PubMed →↩