Melanotan II and Erections: The Accidental Discovery
Melanotan II and Sexual Function
17 of 20 men
In the first human trial, Melanotan II induced penile erections in 17 of 20 men without any sexual stimulation. The peptide had been designed to darken skin. Nobody expected what happened next.
Wessells et al., International Journal of Impotence Research, 2000
Wessells et al., International Journal of Impotence Research, 2000
If you only read one thing
In the 1990s, scientists testing a tanning peptide noticed their male subjects kept getting erections — without any sexual stimulation. That 'side effect' turned out to be a major discovery: the peptide was activating receptors in the brain's sexual arousal center. This led to bremelanotide (Vyleesi), the first FDA-approved drug that actually increases sexual desire rather than just helping with the physical mechanics. It works through the brain, not the blood vessels — which means it can help people who don't respond to Viagra.
In the early 1990s, researchers at the University of Arizona were conducting the first human trials of Melanotan II (MT-II), a cyclic analog of alpha-melanocyte-stimulating hormone (alpha-MSH) designed to induce skin darkening without UV exposure. The goal was photoprotection: a tanning peptide that could reduce skin cancer risk. During these trials, male subjects began reporting spontaneous erections, an effect so unexpected that the researchers initially treated it as a side effect rather than a finding. Mac Hadley, the peptide chemist who helped develop MT-II, later described this as one of the more consequential accidental discoveries in peptide pharmacology.[1]
What followed was a systematic investigation into melanocortin receptors and sexual function that ultimately produced bremelanotide (PT-141), the first centrally-acting peptide drug approved for a sexual dysfunction. The pathway from tanning peptide to sexual medicine illustrates how peptide research produces unexpected findings, and how a single receptor family can govern seemingly unrelated biological processes. For a deeper look at the melanocortin receptor biology connecting these effects, see How Melanocortin Pathways Connect Skin Pigment and Sexual Arousal. For the safety considerations of using unregulated melanotan peptides, see The Risks of Melanotan II for Sexual Enhancement: What You Need to Know.
Key Takeaways
- Scientists were testing a tanning drug when their male subjects kept getting unexpected erections.
- In the first trial, 17 out of 20 men got erections — without any sexual stimulation at all.
- Viagra needs arousal to work. Melanotan generates the arousal signal directly in the brain.
- Among men who didn't respond to Viagra, 34% got working erections on the melanocortin version.
- The side effect became a drug: bremelanotide (Vyleesi) was FDA-approved in 2019 for low female desire.
- It's one of the few sexual-function drugs that actually increases desire, not just the physical mechanics.
- Research-grade Melanotan II still causes nausea, flushing, and yawning — that's why the refined version went to market.
The melanocortin receptor family
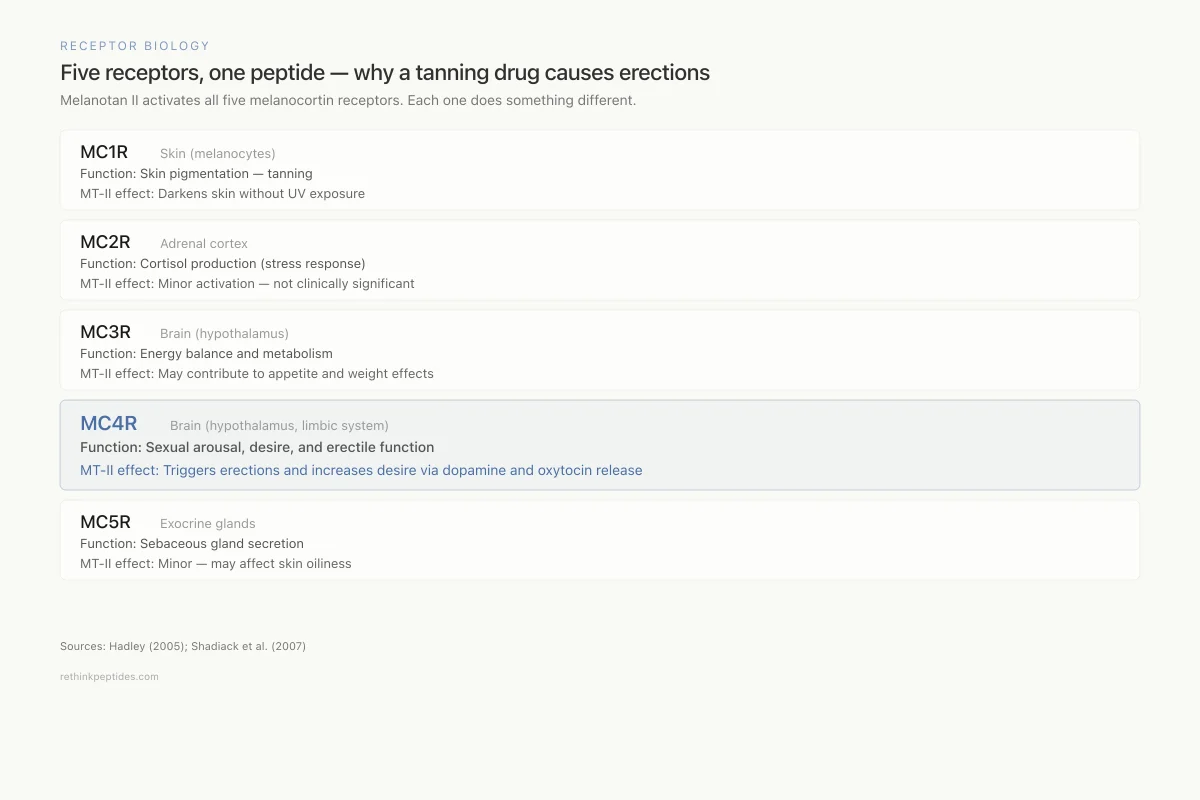
Understanding why a tanning peptide causes erections requires understanding the melanocortin system. The five melanocortin receptors (MC1R through MC5R) are G protein-coupled receptors activated by peptides derived from the precursor protein pro-opiomelanocortin (POMC). Each receptor governs different physiological functions: MC1R controls pigmentation, MC2R mediates adrenal cortisol production, MC3R and MC4R regulate energy balance and sexual function, and MC5R is involved in exocrine gland secretion.[2]
Alpha-MSH, the natural hormone that MT-II was designed to mimic, activates MC1R on melanocytes to stimulate melanin production. But alpha-MSH also activates MC3R and MC4R in the brain. In the 1960s, Ferrari and colleagues had already observed that direct injection of ACTH and alpha-MSH (both POMC-derived peptides) into the brains of experimental animals induced sexual behaviors, including penile erection and increased copulation. These early observations were largely forgotten until the MT-II trials forced researchers to reconsider them.[1]
Receptor Biology
Five receptors, one peptide — why a tanning drug causes erections
Melanotan II activates all five melanocortin receptors. Each one does something different.
MC1R
Skin (melanocytes)Function: Skin pigmentation — tanning
MT-II effect: Darkens skin without UV exposure
MC2R
Adrenal cortexFunction: Cortisol production (stress response)
MT-II effect: Minor activation — not clinically significant at typical doses
MC3R
Brain (hypothalamus)Function: Energy balance and metabolism
MT-II effect: May contribute to appetite and weight effects
MC4R
Brain (hypothalamus, limbic system)Function: Sexual arousal, desire, and erectile function
MT-II effect: Triggers erections and increases sexual desire via dopamine and oxytocin release
MC5R
Exocrine glandsFunction: Sebaceous gland secretion
MT-II effect: Minor — may affect skin oiliness
Source: Hadley (2005); Shadiack et al. (2007)
 View as image
View as imageMT-II is a non-selective melanocortin agonist, meaning it activates all five receptor subtypes. The tanning effect comes from MC1R activation in the skin. The sexual effects come from MC4R activation in the hypothalamus, specifically in the paraventricular nucleus and medial preoptic area, regions that control sexual arousal and erectile function through descending neural pathways to the spinal cord.
The first human trials
Wessells et al. (2000) published the landmark controlled study of MT-II's sexual effects in the International Journal of Impotence Research. Twenty men with no history of erectile dysfunction received subcutaneous MT-II or placebo in a crossover design. The results were striking: erections occurred after 63% of MT-II injections versus 5% of placebo injections. Mean rigidity was 6.9 on a 0-10 scale. The mean duration of tip rigidity greater than 80% (the threshold for penetrative intercourse) was 45 minutes with MT-II compared to 2 minutes for placebo.[3]
These erections occurred in the absence of sexual stimulation. The men were in a clinical research setting being monitored with RigiScan devices. This detail is pharmacologically important: PDE5 inhibitors like sildenafil (Viagra) require sexual stimulation and nitric oxide release to produce an erection. MT-II bypassed this requirement entirely, triggering erectile response through central nervous system activation. The implication was that melanocortins could initiate sexual arousal from the brain downward, rather than facilitating it at the peripheral vascular level.
Hadley (2005) described the broader significance of this finding in a retrospective review. The MT-II data established that melanocortins regulate human sexual function through a centrally-mediated pathway, connecting a system known for controlling pigmentation to one controlling sexual behavior. This was not a minor pharmacological curiosity but a new therapeutic axis for sexual dysfunction.[1]
Discovery Timeline
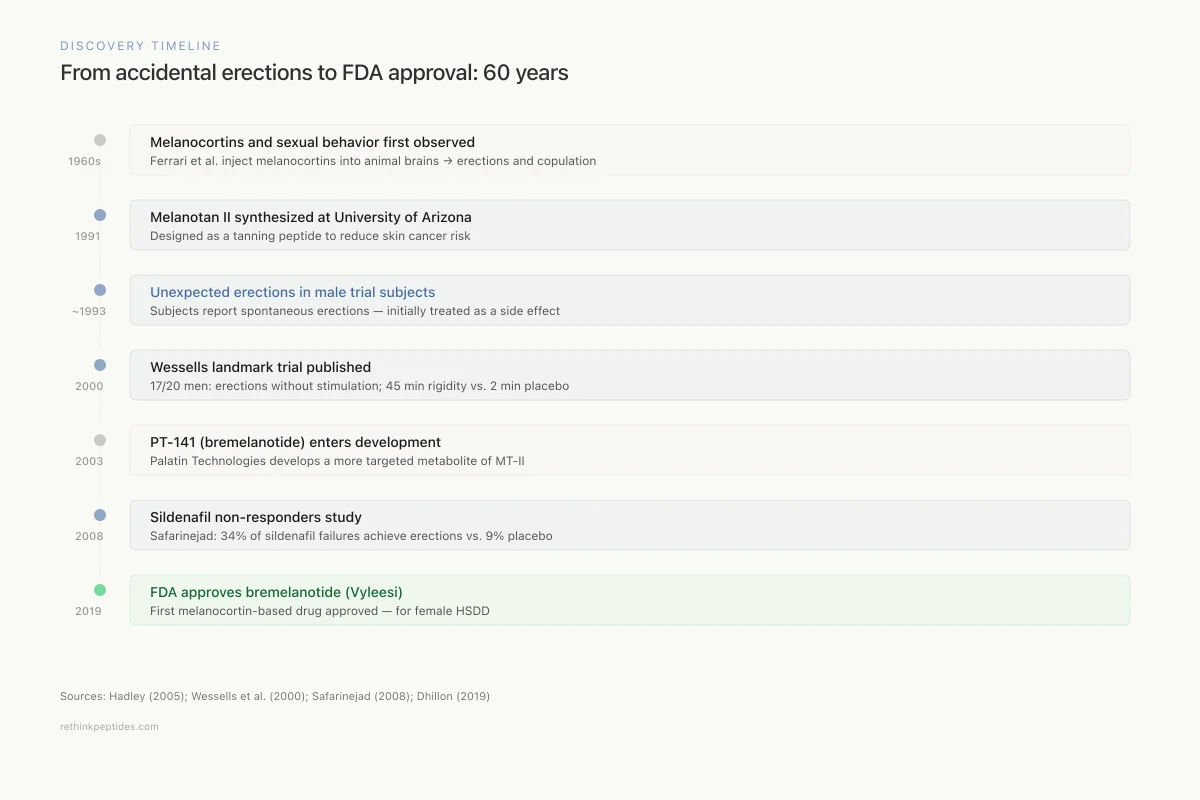
From accidental erections to FDA approval: 60 years of melanocortin sexual medicine
1960s
Melanocortins and sexual behavior first observed
Ferrari et al. inject ACTH/alpha-MSH into animal brains → erections and copulation
1991
Melanotan II synthesized at University of Arizona
Designed as a tanning peptide to reduce skin cancer risk via MC1R activation
~1993
Unexpected erections in male trial subjects
First human MT-II trials: subjects report spontaneous erections — treated as a side effect
2000
Wessells landmark trial published
17 of 20 men developed erections without sexual stimulation; 45 min of >80% rigidity vs. 2 min placebo
2003
PT-141 (bremelanotide) enters development
Palatin Technologies develops a more targeted metabolite of MT-II for sexual dysfunction
2008
Sildenafil non-responders study
Safarinejad: 34% of sildenafil failures achieve erections with bremelanotide vs. 9% placebo
2019
FDA approves bremelanotide (Vyleesi)
First melanocortin-based drug approved — for female HSDD in premenopausal women
Sources: Hadley (2005); Wessells et al. (2000); Dhillon (2019)
 View as image
View as imageThe side effects of MT-II included nausea (the most common), flushing, facial flushing, stretching, and yawning. The nausea was attributed to activation of melanocortin receptors in the brainstem area postrema, a region involved in emesis. The yawning was itself suggestive of central nervous system activity, as yawning is associated with hypothalamic activation and has been linked to sexual arousal in animal models of melanocortin signaling. The combination of tanning, erection, nausea, and yawning from a single peptide reflected MT-II's non-selective activation of melanocortin receptors across multiple organ systems and offered a preview of the pharmacological challenge ahead: how to separate the desired sexual effects from the unwanted peripheral and gastrointestinal effects.
From Melanotan II to bremelanotide (PT-141)
The therapeutic potential of MT-II was clear, but its non-selectivity made it impractical as a drug. It caused skin darkening, nausea, and cardiovascular effects alongside its sexual effects. Palatin Technologies developed PT-141 (later named bremelanotide) as a more targeted compound. PT-141 is a cyclic heptapeptide metabolite of MT-II that retains melanocortin receptor agonist activity but with a modified pharmacological profile.
Molinoff (2003) published the initial pharmacological characterization of PT-141 as a melanocortin agonist for sexual dysfunction treatment.[4] Diamond et al. (2004) conducted the first double-blind, placebo-controlled evaluation of intranasal PT-141 in men, demonstrating dose-dependent erectile responses at doses above 7 mg, with onset approximately 30 minutes after administration.[5]
The intranasal route was initially preferred because it provided rapid absorption and CNS delivery. However, concerns about blood pressure elevation with intranasal dosing led to a switch to subcutaneous injection for clinical development. Diamond et al. (2005) showed that combining low-dose intranasal PT-141 with low-dose sildenafil produced additive effects on erectile function, suggesting that the melanocortin and PDE5 pathways are complementary rather than redundant.[6]
Erectile dysfunction: the sildenafil failure population
Safarinejad (2008) conducted the study that demonstrated bremelanotide's most clinically relevant niche: patients who had failed PDE5 inhibitor therapy. In a randomized, double-blind, placebo-controlled trial of 342 men with erectile dysfunction who did not respond to sildenafil, bremelanotide administered subcutaneously enabled 34% of treated subjects to achieve erections sufficient for intercourse, compared to 9% for placebo.[7]
Mechanism Comparison
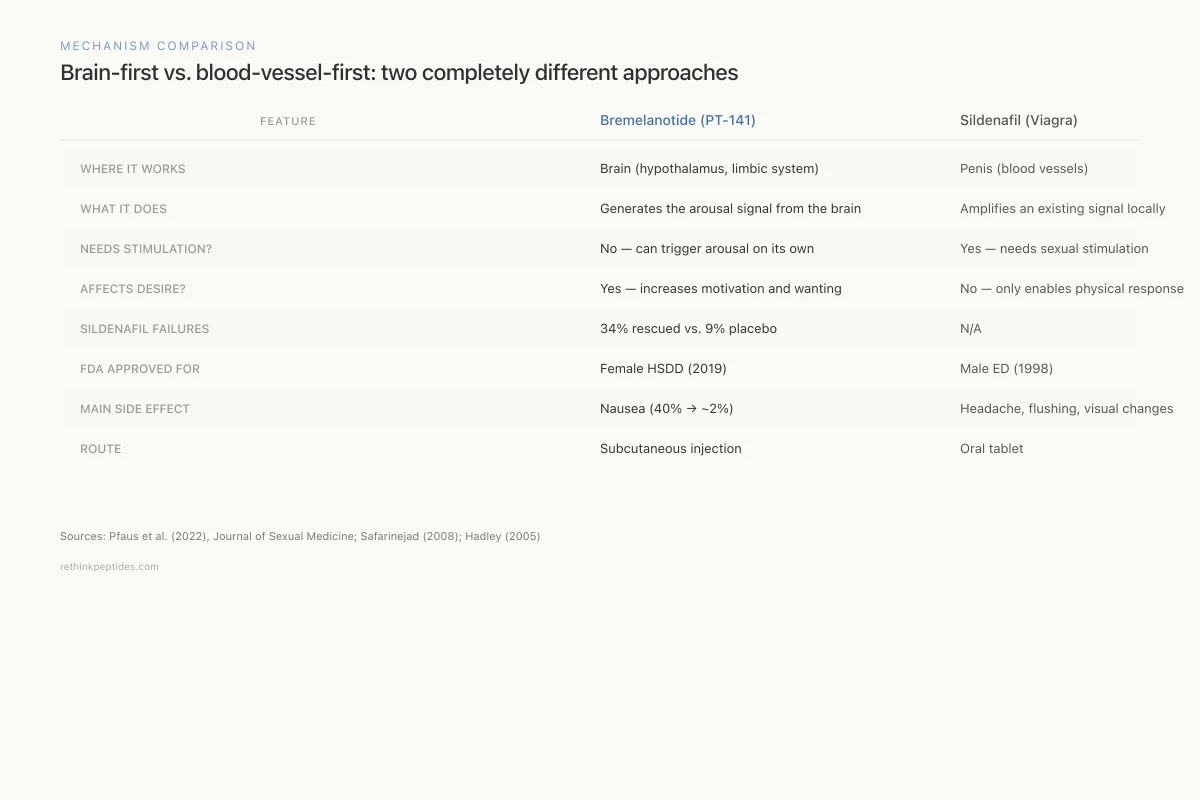
Brain-first vs. blood-vessel-first: two completely different approaches
Bremelanotide (PT-141)
Sildenafil (Viagra)
Where it works
Brain (hypothalamus, limbic system)
Penis (blood vessels)
What it does
Generates the arousal signal from the brain
Amplifies an existing arousal signal locally
Requires sexual stimulation?
No — can trigger arousal on its own
Yes — needs sexual stimulation to work
Affects desire?
Yes — increases motivation and wanting
No — only enables the physical response
Works in sildenafil failures?
Yes — 34% vs. 9% placebo (Safarinejad 2008)
N/A
FDA approved for
Female HSDD (2019)
Male ED (1998)
Main side effect
Nausea (40% initially, drops to ~2%)
Headache, flushing, visual changes
Route
Subcutaneous injection
Oral tablet
Where it works
Bremelanotide: Brain (hypothalamus, limbic system)
Sildenafil: Penis (blood vessels)
What it does
Bremelanotide: Generates the arousal signal from the brain
Sildenafil: Amplifies an existing arousal signal locally
Requires sexual stimulation?
Bremelanotide: No — can trigger arousal on its own
Sildenafil: Yes — needs sexual stimulation to work
Affects desire?
Bremelanotide: Yes — increases motivation and wanting
Sildenafil: No — only enables the physical response
Works in sildenafil failures?
Bremelanotide: Yes — 34% vs. 9% placebo (Safarinejad 2008)
Sildenafil: N/A
FDA approved for
Bremelanotide: Female HSDD (2019)
Sildenafil: Male ED (1998)
Main side effect
Bremelanotide: Nausea (40% initially, drops to ~2%)
Sildenafil: Headache, flushing, visual changes
Route
Bremelanotide: Subcutaneous injection
Sildenafil: Oral tablet
Sources: Pfaus et al. (2022); Safarinejad (2008); Hadley (2005)
 View as image
View as imageThis finding was clinically important because PDE5 inhibitor non-responders represent a substantial population with limited options. Sildenafil and its congeners fail in approximately 30-40% of men with erectile dysfunction, particularly those with severe vascular disease, diabetes-related neuropathy, or radical prostatectomy. Because bremelanotide acts through a centrally-mediated mechanism rather than peripheral vasodilation, it can produce erections even when the local nitric oxide/PDE5 pathway is impaired. This is a fundamental pharmacological distinction: PDE5 inhibitors amplify an existing signal (nitric oxide released during arousal), while melanocortin agonists generate the signal itself from the central nervous system. For patients with vascular damage, nerve damage from surgery, or diabetic neuropathy that disrupts the peripheral arousal pathway, a centrally-acting agent that can initiate the cascade from the brain represents a qualitatively different therapeutic approach.
Uckert et al. (2014) reviewed melanocortin receptor agonists for both male and female sexual dysfunction, noting that the central mechanism of action also meant that melanocortins affected subjective desire and motivation, not just the mechanical ability to achieve erection.[8] This distinction between desire and function became central to bremelanotide's eventual clinical path.
Female sexual dysfunction: where bremelanotide found its market
While the discovery came from male erectile effects, bremelanotide's FDA approval came for female hypoactive sexual desire disorder (HSDD).
Diamond et al. (2006) published one of the first studies showing that PT-141 affected subjective sexual response in women with sexual arousal disorder. Premenopausal women receiving intranasal PT-141 reported increased genital arousal and sexual desire compared to placebo.[9] Pfaus et al. (2007) provided preclinical evidence that bremelanotide facilitated female sexual solicitation in rats through CNS melanocortin pathways.[10]
Kingsberg et al. (2019) published the Phase 3 RECONNECT studies that led to FDA approval. In two randomized, double-blind trials involving over 1,200 premenopausal women with HSDD, subcutaneous bremelanotide (1.75 mg) self-administered before anticipated sexual activity increased the number of satisfying sexual events by approximately 0.5 per month over placebo and reduced distress associated with low sexual desire.[11]
Dhillon (2019) summarized bremelanotide's first approval, marketed as Vyleesi, for HSDD in premenopausal women. It became the second drug approved for this indication (after flibanserin/Addyi) and the first that could be used on-demand rather than daily.[12] Simon et al. (2019) reported long-term safety data showing that bremelanotide's effects were maintained over 12 months, with nausea as the most common adverse event (40% initially, decreasing to approximately 2% with repeated use).[13]
The approval was not without controversy. The effect size in the RECONNECT trials was modest by any measure: an increase of roughly 0.5 satisfying sexual events per month over placebo. Whether this constitutes a clinically meaningful improvement in a condition defined by subjective distress is a question that cuts to the heart of how sexual dysfunction is defined and treated.
Spielmans (2021) published a reanalysis of the Phase 3 data questioning whether the magnitude of benefit justified approval for a condition whose diagnostic validity some researchers dispute. The reanalysis argued that the trials used co-primary endpoints (satisfying sexual events and desire domain scores) that were vulnerable to expectation effects in a condition where the placebo response is characteristically large. Mintzes et al. (2021) similarly criticized the regulatory precedent, arguing that both bremelanotide and flibanserin were approved with effect sizes too small to be clinically meaningful, and that the pharmaceutical industry had created a market by medicalizing normal variations in sexual desire.[14]
The counterargument, articulated by the clinical investigators, is that women with HSDD report genuine distress about absent sexual desire, and that even modest pharmacological improvement can be meaningful when no better options exist. The debate remains active and reflects broader tensions in sexual medicine between patient-reported outcomes, objective effect sizes, and regulatory standards.
The neurobiology: why melanocortins affect desire
Pfaus et al. (2022) published a comprehensive review of bremelanotide's neurobiology in the Journal of Sexual Medicine. The melanocortin system's sexual effects are mediated primarily through MC4R in the hypothalamic paraventricular nucleus and the limbic system, including the amygdala and ventral tegmental area. Activation of MC4R in these regions triggers downstream release of oxytocin and dopamine, neurotransmitters directly involved in sexual motivation and arousal.[15]
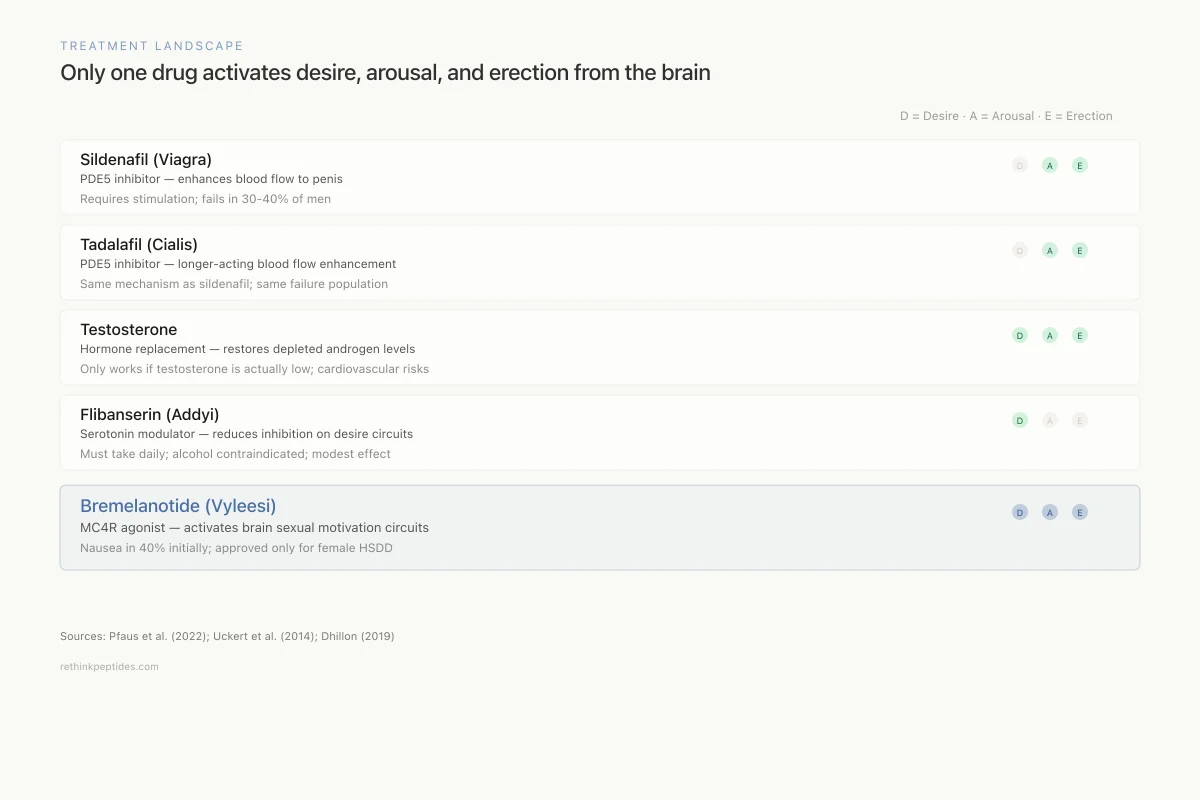
This mechanism distinguishes melanocortin-based treatments from all existing sexual dysfunction drugs. PDE5 inhibitors (sildenafil, tadalafil) facilitate erections by enhancing nitric oxide-mediated blood flow. They do not affect desire. Flibanserin modulates serotonin receptors to reduce inhibitory drive on sexual desire. Testosterone replacement addresses hormonal deficiency. Bremelanotide is the only approved treatment that activates a specific pro-sexual neuropeptide pathway, directly stimulating the brain circuits that generate sexual motivation.
Shadiack et al. (2007) had earlier noted that melanocortins act on both the appetitive (desire/motivation) and consummatory (arousal/orgasm) phases of the sexual response in both sexes.[2] This dual action on desire and arousal is pharmacologically unusual and reflects the melanocortin system's deep evolutionary role in motivational behavior.
Treatment Landscape
Only one drug activates desire, arousal, and erection from the brain
Sildenafil (Viagra)
PDE5 inhibitor — enhances blood flow to penis
Requires sexual stimulation; fails in 30-40% of men
Tadalafil (Cialis)
PDE5 inhibitor — longer-acting blood flow enhancement
Same mechanism as sildenafil; same failure population
Testosterone
Hormone replacement — restores depleted androgen levels
Only works if testosterone is actually low; cardiovascular risks
Flibanserin (Addyi)
Serotonin modulator — reduces inhibition on desire circuits
Must take daily; alcohol contraindicated; modest effect
Bremelanotide (Vyleesi)
MC4R agonist — activates brain sexual motivation circuits
Nausea in 40% initially; approved only for female HSDD
Sources: Pfaus et al. (2022); Uckert et al. (2014); Dhillon (2019)
 View as image
View as imageSafety concerns and the gray market
The story of Melanotan II has a parallel narrative outside the clinical development pipeline. MT-II has been available on the gray market for peptides for over a decade, sold online as a "research chemical" and self-administered by users seeking tanning, sexual enhancement, or both. This unregulated use has produced a case report literature documenting serious adverse events.
Nelson et al. (2012) reported a case of systemic toxicity and rhabdomyolysis following Melanotan II injection purchased online.[16] Ong et al. (2012) described a case of melanoma in situ associated with Melanotan use, raising concern that chronic MC1R stimulation could promote melanocytic neoplasia.[17] The causal link between melanotan and melanoma is not established (the patient may have developed melanoma regardless), but the theoretical concern is biologically plausible: MC1R activation stimulates melanocyte proliferation alongside melanin production.
Safety
ModerateGray-market Melanotan II: documented harm
Concern
Melanotan II purchased online has caused systemic toxicity and rhabdomyolysis (Nelson 2012). A case of melanoma in situ was associated with melanotan use (Ong 2012). Gray-market products have unknown purity, no sterility guarantees, and no dosing standardization.
What the research says
FDA-approved bremelanotide (Vyleesi) has a known safety profile: nausea starts at 40% but drops to ~2% with repeated use. It's the same mechanism but with pharmaceutical-grade manufacturing and medical monitoring.
Particularly relevant for: Anyone purchasing melanotan II from unregulated online sources for tanning or sexual enhancement
What to do
If you're interested in the sexual effects of melanocortins, bremelanotide is available by prescription. Self-injecting gray-market MT-II means unknown purity, no dose standardization, and potential long-term risks to melanocytes.
Nelson et al. (2012); Ong et al. (2012)
Gray-market MT-II products carry additional risks: unknown purity, bacterial contamination from non-sterile manufacturing, incorrect dosing, and no medical monitoring. The gap between the compounded peptide safety monitoring available through legitimate pharmacy channels and the complete absence of oversight for research chemical vendors makes self-administration of MT-II a fundamentally different risk profile from taking FDA-approved bremelanotide under medical supervision.
What the evidence shows and what it does not
The melanocortin sexual pathway is real and well-characterized. MT-II produces erections through MC4R-mediated CNS activation. Bremelanotide, its refined derivative, has demonstrated efficacy in both male erectile dysfunction (particularly in PDE5 non-responders) and female HSDD. The mechanism is fundamentally different from existing treatments, acting on desire and motivation through hypothalamic and limbic circuits rather than on peripheral vascular mechanics.
The limitations are also real. Bremelanotide's approved indication is narrow (premenopausal HSDD only), its effect size in Phase 3 trials was modest, nausea affects a large proportion of initial users, and the blood pressure concerns that derailed the intranasal formulation remain a monitoring issue with subcutaneous injection. The male erectile dysfunction indication, where the early data was most dramatic, has not been pursued to FDA approval. The research showing 34% efficacy in sildenafil non-responders (Safarinejad 2008) has not been followed by Phase 3 trials in this population.
The broader significance lies in what the accidental discovery revealed about biology. A receptor family known for controlling pigmentation also controls sexual behavior, appetite, inflammation, and adrenal function. The melanocortin system is one of the most pleiotropic peptide signaling networks in the body, and the MT-II story is a case study in how peptide research generates insights that extend far beyond the original research question.
The melanocortin sexual pathway also raises questions about what constitutes legitimate drug development versus off-label pharmacological exploration. MT-II remains widely available online as a research peptide, used by thousands of people for tanning and sexual enhancement without medical supervision, while bremelanotide, the FDA-approved refinement, is prescribed for a narrow indication at a cost that limits access. This divergence between unregulated use and regulated medicine is a recurring pattern in peptide pharmacology, similar to the situation with BPC-157 and the FDA, where an unapproved peptide has a widespread user base alongside limited regulatory-pathway development.
Whether future melanocortin-based drugs will address the male sildenafil-failure population, expand to other indications, or achieve the selectivity needed to separate sexual effects from pigmentation and gastrointestinal effects remains to be determined. The biology supports the potential. The clinical and commercial path forward is less clear.
The Bottom Line
Melanotan II, a tanning peptide, accidentally revealed the melanocortin system's role in sexual function when male trial subjects developed spontaneous erections. This discovery led to bremelanotide (PT-141), the first centrally-acting peptide drug for sexual dysfunction, FDA-approved in 2019 for female hypoactive sexual desire disorder. The mechanism operates through MC4R in the hypothalamus and limbic system, stimulating dopamine and oxytocin release to increase sexual desire and arousal. Bremelanotide also showed efficacy in men who failed sildenafil, though this indication was never pursued to approval. Gray-market use of Melanotan II carries documented safety risks. The effect sizes in approved indications are modest, and the full clinical potential of melanocortin-based sexual medicine remains incompletely explored.
Sources & References
- 1RPEP-01041·Hadley, Mac E (2005). “The Accidental Discovery That a Tanning Peptide Enhances Sexual Function in Both Sexes.” Peptides.Study breakdown →PubMed →↩
- 2RPEP-01288·Shadiack, Annette M et al. (2007). “Melanocortins for Both Male AND Female Sexual Dysfunction: PT-141 and Beyond.” Current topics in medicinal chemistry.Study breakdown →PubMed →↩
- 3RPEP-00633·Wessells, H et al. (2000). “Melanocortin receptor agonists, penile erection, and sexual motivation: human studies with Melanotan II..” International journal of impotence research.Study breakdown →PubMed →↩
- 4RPEP-00849·Molinoff, P B et al. (2003). “PT-141: A Melanocortin Peptide That Treats Sexual Dysfunction Through the Brain.” Annals of the New York Academy of Sciences.Study breakdown →PubMed →↩
- 5RPEP-00877·Unknown (2004). “Diamond 2004 Doubleblind Placebocontrolled Evaluation Off.” .Study breakdown →↩
- 6RPEP-01028·Diamond, L E et al. (2005). “Combining Low-Dose PT-141 Nasal Spray With Viagra: Synergistic Erectile Effect.” Urology.Study breakdown →PubMed →↩
- 7RPEP-01413·Safarinejad, Mohammad Reza et al. (2008). “Bremelanotide Rescues Men Who Fail Viagra: A Brain Approach When Blood Flow Drugs Don't Work.” The Journal of urology.Study breakdown →PubMed →↩
- 8RPEP-02565·Ückert, Stefan et al. (2014). “Melanocortin Peptide Agonists Show Promise for Treating Sexual Dysfunction in Both Men and Women Through Central Brain Pathways.” Expert opinion on investigational drugs.Study breakdown →PubMed →↩
- 9RPEP-01128·Diamond, Lisa E et al. (2006). “PT-141 (Bremelanotide) Improves Sexual Arousal in Women With Sexual Arousal Disorder.” The journal of sexual medicine.Study breakdown →PubMed →↩
- 10RPEP-01279·Pfaus, James et al. (2007). “Bremelanotide (PT-141) for Female Sexual Dysfunction: Comprehensive Preclinical CNS Mechanism Review.” The journal of sexual medicine.Study breakdown →PubMed →↩
- 11RPEP-04281·Kingsberg, Sheryl A et al. (2019). “Bremelanotide for the Treatment of Hypoactive Sexual Desire Disorder: Two Randomized Phase 3 Trials..” Obstetrics and gynecology.Study breakdown →PubMed →↩
- 12RPEP-04148·Dhillon, Sohita et al. (2019). “Bremelanotide: First Approval..” Drugs.Study breakdown →PubMed →↩
- 13RPEP-04485·Simon, James A et al. (2019). “Long-Term Safety and Efficacy of Bremelanotide for Hypoactive Sexual Desire Disorder..” Obstetrics and gynecology.Study breakdown →PubMed →↩
- 14RPEP-05614·Mintzes, Barbara et al. (2021). “Bremelanotide and flibanserin for low sexual desire in women: the fallacy of regulatory precedent..” Drug and therapeutics bulletin.Study breakdown →PubMed →↩
- 15RPEP-06431·Pfaus, James G et al. (2022). “The neurobiology of bremelanotide for the treatment of hypoactive sexual desire disorder in premenopausal women..” CNS spectrums.Study breakdown →PubMed →↩
- 16RPEP-02022·Nelson, Michael E et al. (2012). “Melanotan II injection resulting in systemic toxicity and rhabdomyolysis..” Clinical toxicology (Philadelphia.Study breakdown →PubMed →↩
- 17RPEP-02028·Ong, Suyin et al. (2012). “Melanotan-associated melanoma in situ..” The Australasian journal of dermatology.Study breakdown →PubMed →↩