CCK: The First Satiety Peptide Discovered
CCK and Satiety Peptides
1928 Year Discovered
Cholecystokinin was identified by Ivy and Oldberg in 1928 as a hormone that contracts the gallbladder. It became the first gut peptide proven to suppress appetite in 1973, establishing the field of peptide-mediated satiety signaling.
Ivy & Oldberg, American Journal of Physiology, 1928
Ivy & Oldberg, American Journal of Physiology, 1928
If you only read one thing
Cholecystokinin (CCK) is the gut hormone that told science how appetite really works. When you eat fat or protein, cells in your small intestine release CCK. It tells your gallbladder to squeeze, your pancreas to release enzymes, and your brain to stop eating. Scientists have known about it since 1928, and it's the model every modern obesity drug was built against. CCK itself failed as a weight-loss drug because your body adapts to it in days.
Cholecystokinin is the peptide that started the science of gut-mediated satiety. Discovered in 1928 as a gallbladder-contracting hormone, CCK became the first gut peptide proven to reduce food intake in 1973 when James Gibbs and colleagues showed that injecting CCK into rats terminated their meals early. That single experiment launched an entire field. Every peptide-based appetite suppressant that followed, from amylin to GLP-1 to PYY, was studied within a framework CCK established: gut-derived hormones signal the brain through specific receptor systems and vagal pathways to regulate how much you eat.[1]
Today, CCK remains one of the best-characterized satiety peptides in biology. Its two receptor subtypes, CCK-1 (peripheral) and CCK-2 (central), mediate functions spanning digestion, appetite, pain modulation, anxiety, and memory. A 2022 review described CCK as a "master regulator" of upper gastrointestinal function, coordinating gallbladder contraction, pancreatic enzyme secretion, gastric emptying, and meal termination through a single signaling system.[2]
Key Takeaways
- CCK was the first gut peptide proven to suppress appetite (Gibbs et al., 1973), establishing the field of peptide-mediated satiety research
- CCK is released from I-cells in the duodenum in response to dietary fat and protein, activating vagal afferent neurons through CCK-1 receptors to signal meal termination
- CCK-knockout mice are hyperphagic and obese, confirming CCK's role as a physiological satiety signal (Lo et al., 2010)
- The CCK-4 fragment induces panic attacks in 100% of patients with panic disorder and 47% of healthy controls, establishing CCK-2 receptors as targets in anxiety research
- CCK operates on a meal-by-meal basis with rapid tolerance, which is why CCK alone has not produced sustained weight loss in clinical trials
- Modern obesity drugs (GLP-1 agonists, amylin analogs) work through CCK-adjacent pathways; CCK research provided the conceptual framework for all of them
The Discovery: From Gallbladder to Satiety
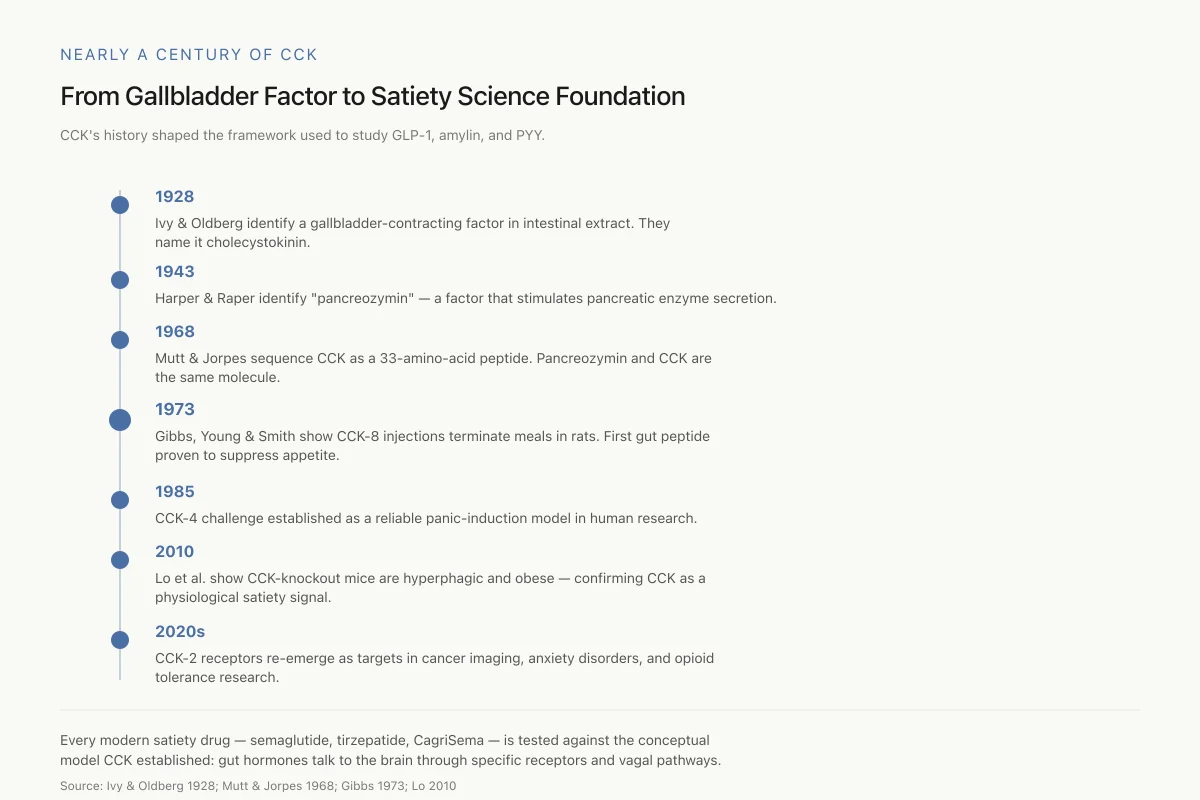
Nearly a Century of CCK
From Gallbladder Factor to Satiety Science Foundation
CCK isn’t just old — its history shaped the entire framework used to study GLP-1, amylin, and PYY.
1928 is ancient for pharmacology. Yet every modern satiety drug — semaglutide, tirzepatide, CagriSema — is tested against the conceptual model CCK established: gut hormones talk to the brain through specific receptors and vagal pathways.
Source: Ivy & Oldberg 1928; Mutt & Jorpes 1968; Gibbs 1973; Lo 2010
 View as image
View as imageThe story of CCK begins with a question about bile. In 1928, Andrew Conway Ivy and Eric Oldberg at Northwestern University Medical School were studying what triggered gallbladder contraction after a meal. They found that intestinal extracts caused the gallbladder to empty and named the responsible factor "cholecystokinin," from the Greek: chole (bile), cyst (bladder), kinin (to move).
For four decades, CCK remained primarily a digestive hormone. In 1943, Harper and Raper identified a factor they called "pancreozymin" that stimulated pancreatic enzyme secretion. It took until 1968 for Viktor Mutt and Erik Jorpes in Stockholm to isolate and sequence CCK from porcine intestine, revealing it as a 33-amino-acid peptide. Their work also showed that cholecystokinin and pancreozymin were the same molecule, consolidating two decades of parallel research.
The molecular biology of CCK became clearer with subsequent work. CCK exists in multiple bioactive forms, all sharing the same C-terminal octapeptide sequence. The major circulating forms include CCK-58, CCK-33, CCK-22, and CCK-8. The sulfated tyrosine at position 7 from the C-terminus is essential for CCK-1 receptor binding and therefore for the digestive and satiety effects. Remove the sulfate group and the peptide loses its ability to contract the gallbladder or suppress appetite.
The satiety connection came in 1973. Gibbs, Young, and Smith demonstrated that intraperitoneal injections of CCK-8 (the eight-amino-acid C-terminal fragment) reduced meal size in rats in a dose-dependent manner. The effect was specific to meal termination, not general malaise: the animals stopped eating sooner but ate normally at the next meal. This was the first experimental proof that a gut-derived peptide could directly suppress appetite through a physiological mechanism. The 1995 characterization of CCK secretion regulation confirmed that fat and protein are the primary dietary triggers, with carbohydrates producing minimal CCK release.[3]
How CCK Works: Receptors and Pathways
One Peptide, Two Receptor Systems
CCK-1 Handles Meals. CCK-2 Handles Fear.
The same peptide can tell your gut to stop eating and your amygdala to panic — the job depends entirely on which receptor it hits.
CCK-1 (formerly CCK-A)
Peripheral / satiety arm
- Gut epithelium, vagal nerve endings, brainstem
- Triggers gallbladder contraction
- Stimulates pancreatic enzyme release
- Slows gastric emptying
- Signals meal termination via vagus → NTS
CCK-2 (formerly CCK-B)
Central / affective arm
- Cortex, amygdala, hippocampus, stomach
- Mediates anxiety and panic responses
- Modulates memory in hippocampal interneurons
- Opposes opioid analgesia (anti-opioid effect)
- Regulates gastric acid secretion
CCK-4, a tiny four-amino-acid fragment, triggers full panic attacks in 100% of panic disorder patients and 47% of healthy controls via CCK-2 — the same molecule whose longer form quietly ends your lunch.
Source: Dockray 2009; Sobrido-Cameán 2020
 View as image
View as imageCCK exerts its effects through two G-protein-coupled receptor subtypes with distinct tissue distributions and functions.
CCK-1 Receptors (Peripheral Satiety)
CCK-1 receptors (formerly called CCK-A, for "alimentary") are concentrated in the gastrointestinal tract, vagal afferent neurons, and specific brain regions including the area postrema and nucleus of the solitary tract. These are the receptors responsible for CCK's satiety effects.[1]
When you eat a meal containing fat or protein, enteroendocrine I-cells in the duodenal and jejunal mucosa release CCK into the bloodstream and the intestinal lumen. The released CCK binds to CCK-1 receptors on vagal afferent nerve endings in the intestinal wall, triggering action potentials that travel up the vagus nerve to the nucleus of the solitary tract in the brainstem.[3] From there, signals project to the hypothalamus and other brain regions involved in appetite regulation.
The satiety signal involves multiple mechanisms operating simultaneously. CCK slows gastric emptying, prolonging gastric distension. It contracts the pyloric sphincter, delaying nutrient transit into the small intestine. It stimulates pancreatic enzyme secretion and gallbladder contraction, facilitating nutrient digestion. And it directly activates vagal satiety pathways. The result is a coordinated "stop eating" signal calibrated to the nutrient content of the meal.
A 2016 study demonstrated that the membrane microenvironment of the CCK-1 receptor modulates its signaling efficiency, meaning that the lipid composition of the cell membrane itself influences how strongly CCK suppresses appetite.[4] This finding added a layer of complexity: CCK sensitivity varies not just with receptor expression levels but with the biophysical properties of the cells expressing those receptors.
CCK-2 Receptors (Brain and Anxiety)
CCK-2 receptors (formerly CCK-B, for "brain") are widely distributed throughout the central nervous system, with high concentrations in the cerebral cortex, amygdala, hippocampus, and brainstem. They are also present in the stomach, where they regulate gastric acid secretion. A 2020 review mapped CCK's central nervous system distribution, documenting its presence in circuits governing anxiety, memory, reward, and nociception.[5]
The most striking demonstration of CCK-2 function is the panic response. CCK-4, a four-amino-acid C-terminal fragment, is one of the most reliable panicogenic agents known. Injected intravenously at doses as low as 25 micrograms, CCK-4 induces panic attacks meeting full DSM-IV criteria in 100% of patients with panic disorder and approximately 47% of healthy controls. The attacks are rapid-onset, peaking within 30 to 60 seconds of injection, and include tachycardia, dyspnea, derealization, and intense fear.
This CCK-panic link has made CCK-4 challenge a standard research model for testing anxiolytic drugs. A 2021 study provided evidence that increased cholecystokinin signaling in the periaqueductal gray matter contributes to threat-related behavioral responses, further connecting CCK-2 activation to the neural architecture of fear and panic.[6]
A 2025 study mapped neuronal CCK populations across brain regions, identifying distinct CCK-expressing circuits involved in feeding behavior, anxiety-like responses, and reward processing.[7] The finding that the same peptide can simultaneously suppress appetite (through CCK-1 receptors in the gut-brain axis) and modulate fear (through CCK-2 receptors in limbic circuits) illustrates the complexity of peptide signaling systems. These are not simple on-off switches. They are context-dependent modulators whose effects depend entirely on where they act and which receptor they bind.
Safety
CriticalCCK-4 is a research-only panicogen
Concern
CCK-4 injection triggers full DSM-criteria panic attacks in 100% of panic disorder patients and ~47% of healthy volunteers within 30–60 seconds — tachycardia, dyspnea, derealization, intense fear.
What the research says
CCK-4 is used only in controlled research settings with informed consent and monitoring. It is not a clinical agent and should never be obtained outside formal studies.
Particularly relevant for: Anyone who encounters CCK-4 outside of a sanctioned clinical trial
What to do
Do not self-administer CCK-4. It is a reliable panic inducer, not a therapeutic.
Keay 2021; published CCK-4 challenge literature
CCK and Appetite Regulation: What the Evidence Shows
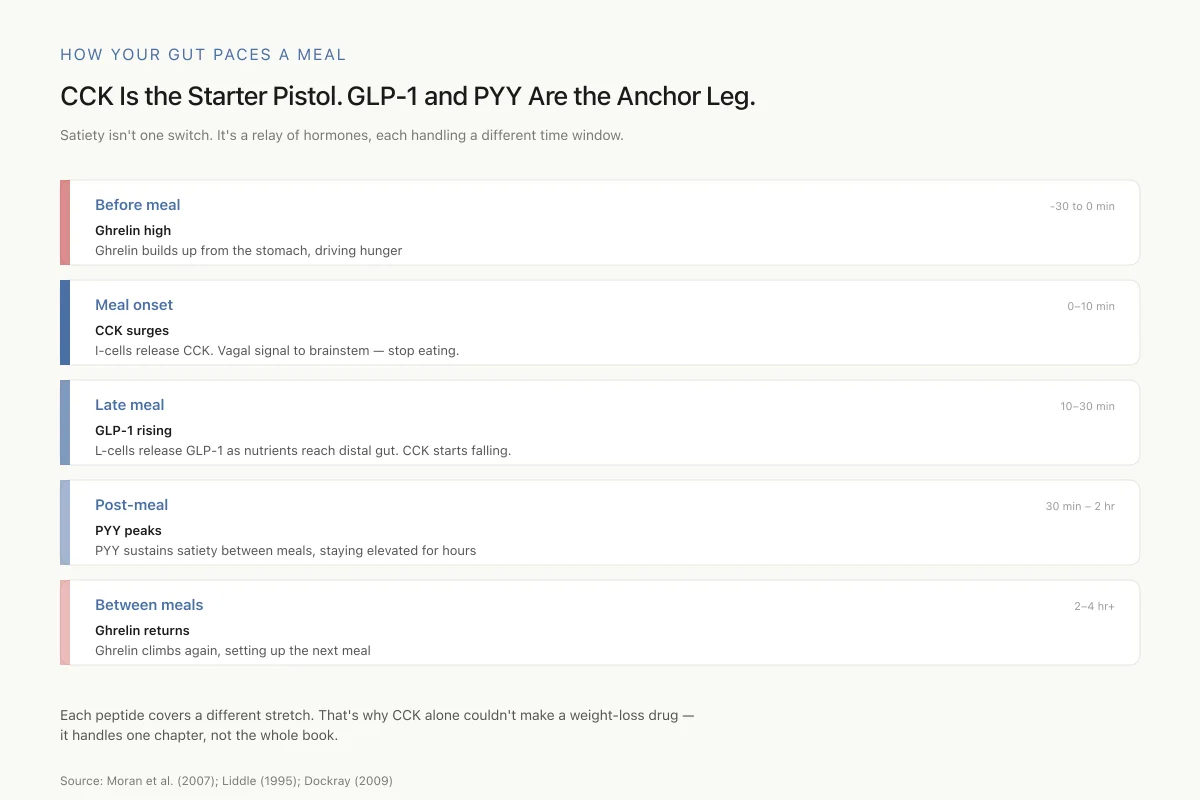
How Your Gut Paces a Meal
CCK Is the Starter Pistol. GLP-1 and PYY Are the Anchor Leg.
Satiety isn’t one switch. It’s a relay of hormones, each handling a different time window.
Ghrelin high
Ghrelin builds up from the stomach, driving hunger
CCK surges
I-cells release CCK in response to fat and protein. Vagal signal to brainstem — stop eating.
GLP-1 rising
L-cells release GLP-1 as nutrients reach distal gut. CCK starts falling as the meal is processed.
PYY peaks
PYY sustains satiety between meals, staying elevated for hours
Ghrelin returns
Ghrelin climbs again, setting up the next meal
Each peptide covers a different stretch. That’s why CCK alone couldn’t make a weight-loss drug — it handles one chapter, not the whole book.
Source: Moran et al. (2007); Liddle (1995); Dockray (2009)
 View as image
View as imageThe foundational question for any satiety peptide is whether it is a true physiological regulator of food intake or merely capable of suppressing appetite when administered as a drug. For CCK, the evidence supports a genuine regulatory role.
CCK-knockout mice are hyperphagic. They eat larger meals, gain more weight, and accumulate more body fat than wild-type controls.[8] This was a critical finding because it confirmed that endogenous CCK, not just exogenous injections, is required for normal meal termination. Without CCK, mice cannot properly gauge when a meal should end.
The interplay between CCK and other appetite hormones adds further complexity. Ghrelin, the "hunger hormone" released from the stomach before meals, suppresses CCK's satiety signal. A 2017 study showed that ghrelin directly inhibits cholecystokinin secretion from intestinal I-cells, creating a competitive dynamic: ghrelin drives you to eat while CCK signals you to stop.[9] The balance between these opposing signals determines meal size. For how CCK coordinates with other gut satiety peptides including GLP-1 and PYY, see How GLP-1, PYY, and CCK Work Together to Stop You from Eating.
A 2007 study measured post-prandial levels of ghrelin, CCK, and peptide YY simultaneously in healthy men, demonstrating the temporal sequence of gut hormone release.[10] CCK rises rapidly within minutes of meal onset and peaks before the meal ends, functioning as an acute meal termination signal. PYY rises more slowly, peaking 1 to 2 hours after the meal and remaining elevated for several hours. Ghrelin, the hunger signal, drops as CCK rises. This creates a relay system: ghrelin drops to remove the drive to eat, CCK surges to terminate the current meal, and PYY sustains satiety into the inter-meal interval. The elegance of this system is that each peptide handles a different time window of appetite regulation.
The practical limitation of CCK for weight management is tolerance. CCK suppresses appetite on a meal-by-meal basis, but its effect diminishes with repeated exposure. Animals given CCK before every meal initially eat less, but within days they compensate by increasing meal frequency. This rapid tolerance is the fundamental reason CCK agonists have not succeeded as standalone obesity drugs. The body treats CCK as a short-term meal termination signal, not a long-term energy balance regulator. Modern obesity pharmacology has solved this problem by targeting peptide systems with longer-acting effects. For the broader story of how amylin-based drugs overcame the tolerance problem, see Cagrilintide: The Amylin Half of CagriSema.
CCK Beyond Appetite: Digestion, Pain, and Memory
CCK's biological roles extend well beyond satiety.
Gallbladder and pancreatic function. CCK's original identity as a digestive hormone remains its most established clinical application. CCK-8 injection (sincalide, marketed as Kinevac) is used diagnostically to assess gallbladder ejection fraction in patients with suspected biliary dyskinesia. The CCK-stimulated HIDA scan remains a standard clinical test: patients receive an intravenous infusion of sincalide while a gamma camera tracks gallbladder emptying. An ejection fraction below 35% is considered abnormal and supports the diagnosis of gallbladder dysfunction. This is one of the few direct clinical uses of any gut peptide as a diagnostic agent.
CCK also stimulates pancreatic enzyme secretion through CCK-1 receptors on pancreatic acinar cells, triggering release of lipases, proteases, and amylases into the duodenum. Impaired CCK signaling has been linked to exocrine pancreatic insufficiency, and CCK levels are used as a biomarker of pancreatic function in some clinical contexts. The dual role of stimulating both gallbladder contraction and pancreatic secretion ensures that bile and digestive enzymes arrive in the duodenum simultaneously to process the fat and protein that triggered CCK release in the first place. It is an elegant feedback loop: nutrients trigger CCK release, CCK triggers the delivery of digestive machinery, and the resulting nutrient absorption eventually reduces CCK secretion as the meal is processed.
Pain modulation. CCK acts as an anti-opioid peptide in the central nervous system. It directly opposes the analgesic effects of endogenous opioids and exogenous opioid drugs by activating CCK-2 receptors in the rostral ventromedial medulla and periaqueductal gray. This is clinically relevant: elevated CCK levels have been associated with the development of opioid tolerance, and CCK receptor antagonists (proglumide, devazepide) have been studied as adjuncts to opioid therapy to prevent tolerance development. The anti-opioid effect of CCK may also explain why some patients develop hyperalgesia during chronic opioid therapy. Blocking CCK-2 receptors in animal models restored opioid sensitivity and reduced tolerance.
Memory and cognition. CCK is co-localized with GABA in hippocampal interneurons, where it modulates synaptic transmission and memory formation. These CCK-expressing basket cells are among the most abundant interneuron types in the hippocampus and cortex. They regulate the balance of excitation and inhibition by controlling the timing of pyramidal cell firing, a process essential for memory encoding and retrieval. Disruption of CCK signaling in these circuits has been associated with cognitive deficits in preclinical models. A 2025 review of neuronal CCK function described it as a "node peptide" connecting metabolic state with cognitive performance, noting that the same peptide that tells you to stop eating also influences how well your hippocampus encodes the experience of that meal.[7] This dual metabolic-cognitive role may explain why CCK levels are altered in some neurodegenerative conditions.
Gut peptide interactions. CCK does not operate in isolation. It interacts with virtually every other gut hormone. It stimulates GLP-1 release through paracrine signaling in the intestinal epithelium. It modulates ghrelin secretion. It potentiates the effects of leptin. A 2025 study identified novel peptides that stimulate CCK secretion from I-cells, suggesting that the CCK regulatory network is even more complex than previously understood.[11] For a broader look at another satiety peptide that works alongside CCK, see Peptide YY (PYY): The "I'm Done Eating" Signal from Your Gut. For the dual-agonist natural hormone that bridges CCK's domain with GLP-1 signaling, see Oxyntomodulin: The Natural Dual Agonist That Suppresses Appetite.
Why CCK Failed as a Weight Loss Drug
The history of CCK-based obesity therapeutics is instructive precisely because it failed. Multiple CCK-1 receptor agonists entered clinical development. None reached the market for weight management.
The problem is not efficacy per meal. CCK reliably reduces meal size. The problem is the body's compensatory response. When CCK shortens one meal, the next meal comes sooner. Daily caloric intake remains stable or returns to baseline within days. This pattern, called compensatory hyperphagia, defeated every CCK agonist tested.
The lesson shaped all subsequent satiety peptide drug development. GLP-1 agonists succeeded where CCK failed because GLP-1's appetite-suppressing effects persist with chronic dosing, likely because GLP-1 acts on both peripheral and central circuits with slower desensitization kinetics. Amylin analogs target a separate satiety circuit in the area postrema that shows less rapid tolerance than the vagal CCK pathway. The modern combination strategies (CagriSema, tirzepatide, retatrutide) layer multiple peptide mechanisms precisely to prevent the kind of compensatory adaptation that CCK monotherapy could not overcome.
There is another reason CCK agonists may have failed. CCK has a very short plasma half-life, approximately 1 to 2 minutes. The peptide is rapidly degraded by endopeptidases. Even synthetic CCK analogs with improved stability could not match the pharmacokinetic profiles of modern peptide drugs with half-lives measured in days (semaglutide: 7 days; cagrilintide: 8 days). The field learned that sustained receptor engagement, not just acute activation, is required for clinically meaningful appetite suppression over weeks and months.
CCK's failure as a drug illuminated a fundamental principle: meal-by-meal satiety signals and long-term energy balance regulation are separate systems. Activating the first does not automatically control the second. That insight, drawn from decades of CCK research, is embedded in every modern obesity therapeutic pipeline. For the broader framework of how modern drugs combine multiple peptide mechanisms, see Pancreatic Polypeptide: The Underappreciated Appetite Regulator.
Where CCK Research Stands Today
CCK research continues to produce findings, though the focus has shifted from drug development to systems biology. The current questions are less about whether CCK can be a weight loss drug and more about how CCK signaling integrates with the broader hormonal network that governs metabolic health.
Active research areas include the role of CCK in the gut-brain axis during neurodegeneration, the connection between impaired CCK signaling and post-bariatric surgery outcomes, and the use of CCK-2 receptor antagonists as psychiatric therapeutics. A 2022 review examining the conservation of CCK/sulfakinin signaling from insects through mammals documented that this peptide system is at least 500 million years old, making it one of the most ancient regulatory mechanisms for nutrient intake and energy allocation.[2]
The revival of interest in CCK receptor pharmacology is also driven by cancer research. CCK-2 receptors are overexpressed in several tumor types, including pancreatic adenocarcinoma, medullary thyroid carcinoma, and certain gastric cancers. Radiolabeled CCK analogs are being developed as targeted imaging agents and potential peptide receptor radionuclide therapy (PRRT) vehicles, following the model established by somatostatin analogs in neuroendocrine tumors. This represents an entirely different therapeutic application for a peptide originally studied for its effects on digestion and appetite.
For the broader landscape of satiety peptides, including how CCK's short-acting signal fits into the longer-duration systems now targeted by drugs, see How GLP-1, PYY, and CCK Work Together to Stop You from Eating.
The Bottom Line
CCK was the first gut peptide proven to suppress appetite, establishing the entire field of peptide-mediated satiety research in 1973. It operates through two receptor subtypes: CCK-1 in the periphery for satiety and digestion, and CCK-2 in the brain for anxiety, pain modulation, and cognition. CCK failed as a weight loss drug because of rapid tolerance, but the principles learned from its failure shaped every modern obesity therapeutic, from GLP-1 agonists to CagriSema.
Sources & References
- 1RPEP-01473·Dockray, Graham J (2009). “CCK and Gut-Brain Signalling: The Complete 2009 Review of the Satiety Hormone's Pathways.” Regulatory peptides.Study breakdown →PubMed →↩
- 2RPEP-06148·Gomes, Ana S et al. (2022). “The role of cholecystokinin and peptide YY in feed intake in Atlantic halibut (Hippoglossus hippoglossus) larvae..” Neuropeptides.Study breakdown →PubMed →↩
- 3RPEP-00326·Liddle, R A (1995). “How Dietary Proteins and Fats Trigger the Release of Digestion-Controlling CCK.” The American journal of physiology.Study breakdown →PubMed →↩
- 4RPEP-02914·Desai, A J et al. (2016). “Cholecystokinin-induced satiety, a key gut servomechanism that is affected by the membrane microenvironment of this receptor..” International journal of obesity supplements.Study breakdown →PubMed →↩
- 5RPEP-05143·Sobrido-Cameán, D et al. (2020). “Ancient Sea Lamprey Brain Reveals Evolutionary Origins of the Gut-Brain Peptide CCK.” Brain structure & function.Study breakdown →PubMed →↩
- 6RPEP-05488·Keay, Kevin A et al. (2021). “Cholecystokinin Peptide in the Brain Explains Why Chronic Pain Disrupts Social Behavior.” Journal of neurochemistry.Study breakdown →PubMed →↩
- 7RPEP-11455·Huang, Feng-Wen et al. (2025). “Role of Neuronal Cholecystokinin Receptor: An Emerging Therapeutic Target for Ameliorating Neurological Diseases..” Molecular neurobiology.Study breakdown →PubMed →↩
- 8RPEP-01653·Lo, Chun-Min et al. (2010). “Mice Without CCK Are Resistant to High-Fat Diet Obesity: The Satiety Hormone's Paradoxical Role.” Gastroenterology.Study breakdown →PubMed →↩
- 9RPEP-03218·Blanco, Ayelén Melisa et al. (2017). “Ghrelin suppresses cholecystokinin (CCK), peptide YY (PYY) and glucagon-like peptide-1 (GLP-1) in the intestine, and attenuates the anorectic effects of CCK, PYY and GLP-1 in goldfish (Carassius auratus)..” Hormones and behavior.Study breakdown →PubMed →↩
- 10RPEP-01269·Moran, Lisa J et al. (2007). “Weight Loss in Overweight Women Changes Gut Hormones — And Not Always Favorably for Keeping Weight Off.” The American journal of clinical nutrition.Study breakdown →PubMed →↩
- 11RPEP-13647·Song, Hongdong et al. (2025). “Oat protein peptides stimulate appetite-suppressing hormone CCK through calcium-sensing receptor signaling.” Journal of agricultural and food chemistry.Study breakdown →PubMed →↩