Sirtuins and Peptides: The Longevity Gene Connection
Sirtuins and Peptide Regulation
7 sirtuin isoforms
Mammals possess seven sirtuin enzymes (SIRT1-7) that depend on NAD+ to regulate gene expression, DNA repair, and metabolic homeostasis across virtually every tissue.
Wan et al., Journal of Translational Medicine, 2023
Wan et al., Journal of Translational Medicine, 2023
If you only read one thing
Sirtuins are a family of seven enzymes that your body uses to repair DNA, manage metabolism, and respond to stress. They all need a fuel called NAD+ to work, and NAD+ drops by half or more as you age — so your sirtuins slow down too. Several peptides, especially the mitochondrial peptide MOTS-c, can boost sirtuin activity by refilling the NAD+ tank. But 'sirtuins = longevity' is an oversimplification — the early hype was based on yeast studies that didn't fully hold up in mammals. The real picture is more nuanced: sirtuins help with healthy aging, but they're part of a bigger system, not a single longevity switch.
Sirtuins were first identified in yeast in the 1970s as the Silent Information Regulator 2 (Sir2) gene, which extended replicative lifespan when overexpressed. Since then, seven mammalian homologs (SIRT1 through SIRT7) have been characterized, all sharing a dependence on nicotinamide adenine dinucleotide (NAD+) as an essential cofactor for their deacetylase and deacylase activity.[1] What makes sirtuin biology relevant to peptide research is accumulating evidence that mitochondrial-derived peptides, synthetic peptide activators, and endogenous peptide hormones intersect with sirtuin signaling at multiple points. MOTS-c activates AMPK upstream of SIRT1. Humanin regulates lifespan through FOXO/daf-16, a pathway sirtuins also govern. GHK-Cu modulates gene expression patterns that overlap with sirtuin-regulated transcriptional networks. This article maps those intersections and examines what the evidence supports, where the gaps remain, and what these peptide-sirtuin connections mean for longevity science. For a deeper look at how NAD+ metabolism specifically intersects with peptide pathways, see Where NAD+ and Peptide Longevity Pathways Intersect.
Key Takeaways
- Sirtuins are a family of seven enzymes that handle DNA repair, metabolism, and stress in every cell.
- Sirtuins need NAD+ to work, and your NAD+ drops by more than half as you age.
- "Sirtuins are longevity genes" is an oversimplification — early yeast findings didn't hold up in mammals.
- Cranking up SIRT6 in male mice did extend life by about 15% in one Nature study.
- Several peptides — MOTS-c, humanin, even Ozempic — all funnel into the sirtuin system through different doors.
- Not every sirtuin is a friend — SIRT4 can actually accelerate heart failure under some conditions.
- The strongest natural boosts to your sirtuins are still the same two things: exercise and eating less.
What Sirtuins Actually Do
Sirtuins remove acetyl groups (and other acyl modifications) from lysine residues on histones and non-histone proteins. This deacetylation requires NAD+ as a co-substrate, which is consumed in the reaction and converted to nicotinamide. The NAD+ dependence is what links sirtuin activity directly to cellular energy status: when NAD+ levels are high (as during caloric restriction or exercise), sirtuin activity increases; when NAD+ drops (as during aging or metabolic dysfunction), sirtuin activity declines.[1]
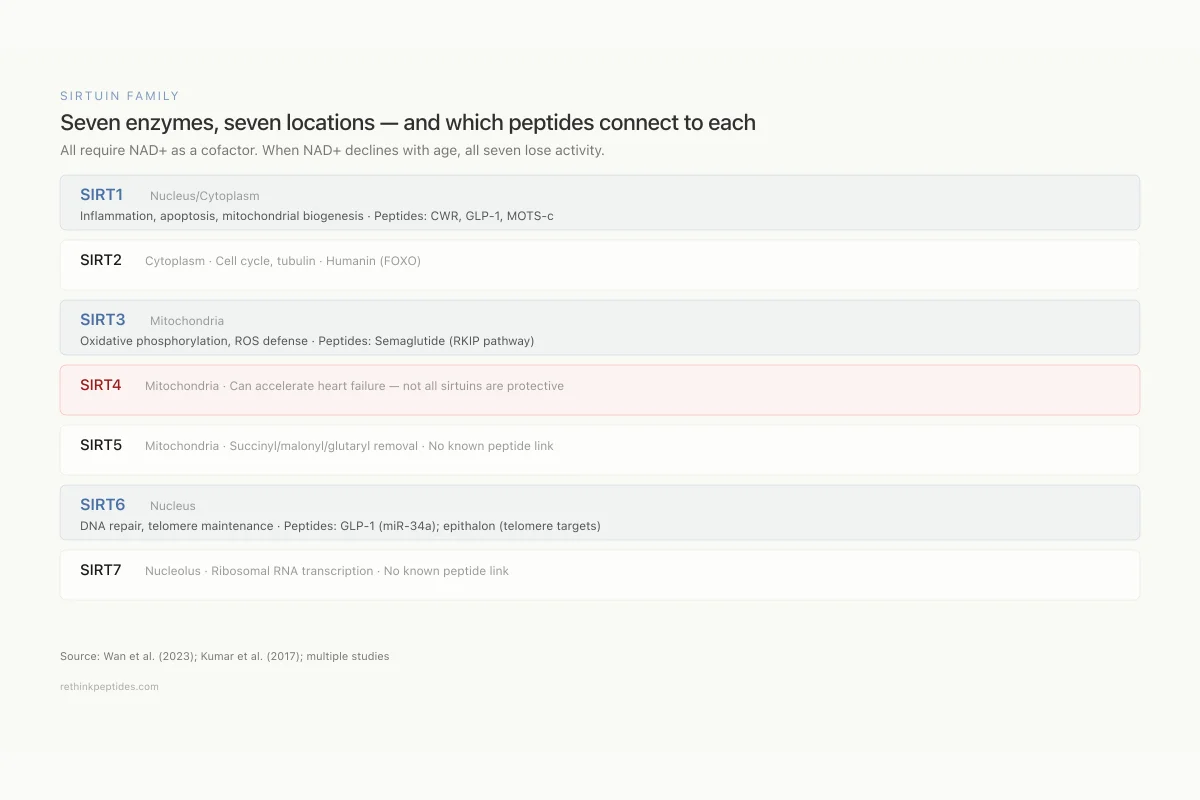
Sirtuin Family
Seven enzymes, seven locations — and which peptides connect to each
All require NAD+ as a cofactor. When NAD+ declines with age, all seven lose activity.
SIRT1
Nucleus / CytoplasmInflammation, apoptosis, mitochondrial biogenesis
Peptide links: CWR tripeptide (direct activator); GLP-1 agonists; MOTS-c (via AMPK)
SIRT2
CytoplasmCell cycle, tubulin regulation
Peptide links: Humanin (via FOXO deacetylation)
SIRT3
MitochondriaOxidative phosphorylation, ROS defense, fatty acid oxidation
Peptide links: Semaglutide (cardiac inflammation via RKIP)
SIRT4
MitochondriaMetabolic regulation — but can accelerate heart failure
Peptide links: No known peptide connection
SIRT5
MitochondriaRemoves succinyl, malonyl, glutaryl modifications
Peptide links: No known peptide connection
SIRT6
NucleusDNA repair, telomere maintenance, glucose homeostasis
Peptide links: GLP-1 agonists (via miR-34a); epithalon (shared telomere targets)
SIRT7
NucleolusRibosomal RNA transcription
Peptide links: No known peptide connection
Source: Wan et al. (2023); Kumar et al. (2017); multiple studies
 View as image
View as imageEach of the seven mammalian sirtuins occupies a different cellular compartment and serves different functions:
- SIRT1 (nucleus/cytoplasm): The most studied isoform. Deacetylates histones, p53, FOXO transcription factors, PGC-1alpha, and NF-kappaB. Regulates inflammation, apoptosis, and mitochondrial biogenesis.
- SIRT2 (cytoplasm): Deacetylates tubulin and regulates cell cycle progression.
- SIRT3 (mitochondria): The primary mitochondrial deacetylase. Regulates oxidative phosphorylation, fatty acid oxidation, and reactive oxygen species (ROS) management. Semaglutide has been shown to protect against cardiac inflammation through SIRT3-dependent RKIP pathway activation.[2]
- SIRT4 (mitochondria): Unlike other sirtuins, SIRT4 has been shown to accelerate heart failure by enhancing ROS-mediated profibrotic signaling, demonstrating that not all sirtuin activity is protective.[3]
- SIRT5 (mitochondria): Removes succinyl, malonyl, and glutaryl modifications.
- SIRT6 (nucleus): Regulates DNA repair, telomere maintenance, and glucose homeostasis. SIRT6 overexpression extended lifespan in male mice. GLP-1 agonists have been shown to activate SIRT6 through miR-34a modulation, reducing vascular calcification in aged rats.[4]
- SIRT7 (nucleolus): Regulates ribosomal RNA transcription.
The Sirtuin-Longevity Debate
The idea that sirtuins are "longevity genes" traces back to Kaeberlein et al. (1999), who showed that extra copies of Sir2 extended yeast replicative lifespan by approximately 30%. Similar findings followed in C. elegans and Drosophila. The narrative was compelling: a single gene family, activated by caloric restriction, that extended lifespan across species.
That narrative has been substantially complicated. A 2011 study by Burnett et al. found that the original worm and fly lifespan extensions were confounded by genetic background effects. When properly controlled, the effects were much smaller or absent. A 2022 analysis in Life Metabolism (Tsuchiya et al.) directly challenged whether sirtuins are conserved longevity genes, noting that most mammalian sirtuin-overexpression studies show health benefits but not consistent lifespan extension. Derek Lowe, writing in Science, described the entire sirtuin-longevity area as resting on a foundation of "artifactual results, overinterpretation, and publication bias."
What the evidence does support: SIRT1 and SIRT6 overexpression in mice produces metabolic improvements resembling caloric restriction. SIRT6-overexpressing male mice lived approximately 15% longer in one study published in Nature (Kanfi et al., 2012). SIRT1-overexpressing mice showed improved glucose tolerance, lower inflammation, and reduced cancer incidence, but lifespan extension was modest and sex-dependent. The field has shifted from "sirtuins cause longevity" to "sirtuins mediate some benefits of caloric restriction and exercise." That distinction matters for understanding how peptides interact with these pathways.
NAD+ Decline: The Rate-Limiting Factor
All seven sirtuins consume NAD+ in their enzymatic reactions. NAD+ levels decline substantially with age: by middle age, tissue levels can be roughly half of youthful concentrations, and by old age the decline may reach 60-80% depending on the tissue. This means that even if sirtuin protein levels remain stable, their activity falls because the essential cofactor is depleted.
This NAD+ decline creates a bottleneck that affects every sirtuin isoform simultaneously. It also provides a mechanistic explanation for why caloric restriction and exercise enhance sirtuin activity: both increase NAD+ biosynthesis through different routes. Caloric restriction activates NAMPT (nicotinamide phosphoribosyltransferase), the rate-limiting enzyme in the NAD+ salvage pathway. Exercise increases NAD+ through AMPK-mediated NAMPT upregulation. Several peptides, particularly MOTS-c, feed into this same AMPK-NAMPT-NAD+ axis, raising the question of whether they can partially compensate for age-related NAD+ decline.
MOTS-c: The Mitochondrial Peptide That Activates Sirtuin Pathways
MOTS-c (Mitochondrial ORF of the Twelve S rRNA type-c) is a 16-amino-acid peptide encoded within the mitochondrial genome. Discovered by Lee et al. in 2015, it was only the second mitochondrial-derived peptide identified after humanin.[5] For a comprehensive overview of MOTS-c research, see MOTS-c: The Mitochondrial Peptide That Mimics Exercise.
MOTS-c connects to sirtuin biology through AMPK. The peptide inhibits the folate cycle and de novo purine biosynthesis, leading to accumulation of AICAR (5-aminoimidazole-4-carboxamide ribonucleotide), which activates AMPK.[6] AMPK, in turn, increases the NAD+/NADH ratio and activates SIRT1. This cascade means MOTS-c acts as an upstream activator of the AMPK-SIRT1 axis without directly binding sirtuins.
Nuclear Translocation Under Stress
In 2018, Kim et al. published a landmark finding in Cell Metabolism: MOTS-c translocates from the cytoplasm to the nucleus under metabolic stress, where it regulates nuclear gene expression. In the nucleus, MOTS-c interacts with stress-responsive transcription factors including NRF2 (nuclear factor erythroid 2-related factor 2) and regulates genes containing antioxidant response elements (AREs).[7] This was the first evidence that a mitochondrial-encoded factor directly controls nuclear gene expression, suggesting the mitochondrial and nuclear genomes co-evolved mechanisms for cross-regulation.
The NRF2 connection is notable because SIRT1 also activates NRF2-dependent antioxidant gene expression through deacetylation. MOTS-c and SIRT1 thus converge on the same transcriptional targets through different mechanisms. A 2026 study confirmed MOTS-c improves muscle mitochondrial bioenergetics through PGC-1alpha/AMPK-dependent pathways, the same pathway axis that SIRT1 regulates through PGC-1alpha deacetylation.[8]
MOTS-c and Exercise
Exercise increases circulating MOTS-c levels in humans. Dieli-Conwright et al. (2021) measured MOTS-c in Hispanic and non-Hispanic White breast cancer survivors undergoing aerobic and resistance exercise and found significant increases in circulating levels.[9] Exercise also activates sirtuins through increased NAD+ availability. The parallel activation of both pathways during exercise suggests they form a coordinated metabolic response rather than operating independently. MOTS-c levels decline with age; a 2025 study found reduced serum MOTS-c in patients with obstructive sleep apnea compared to controls.[10] For more on the intersection of MOTS-c with NAD+ biology, see MOTS-c and NAD+ Metabolism: Mitochondrial Peptides Meet Cellular Energy.
MOTS-c Genetic Variation
A naturally occurring MOTS-c variant (m.1382A>C, resulting in a K14Q amino acid change) is found at higher frequency in East Asian populations and is associated with increased type 2 diabetes risk. Zempo et al. (2021) showed this polymorphism reduces MOTS-c's metabolic protective effects, providing genetic evidence that MOTS-c function directly influences metabolic aging in humans.[11]
Humanin: Lifespan Extension Through Sirtuin-Adjacent Pathways
Humanin is a 24-amino-acid peptide encoded within the mitochondrial 16S rRNA gene. Discovered in 2001 through a functional screen for factors that protect neurons against amyloid-beta toxicity, it has since been shown to regulate lifespan in multiple species. For a full overview, see Humanin: The Cytoprotective Peptide from Your Mitochondria.
The 2020 Yen et al. study provided the most direct evidence linking humanin to longevity. Humanin overexpression extended C. elegans lifespan in a daf-16/FOXO-dependent manner. In mice, twice-weekly injections of the humanin analog HNG in middle-aged animals improved metabolic healthspan parameters and reduced inflammatory markers. Circulating humanin levels were stable in naked mole-rats (a model of negligible senescence) but declined with age in other species. Children of centenarians had significantly higher circulating humanin than age-matched controls.[12]
Where Humanin Meets Sirtuins
The FOXO connection is the key link. Sirtuins (particularly SIRT1 and SIRT2) deacetylate FOXO transcription factors, modulating their activity. In C. elegans, the sirtuin homolog Sir2.1 regulates lifespan partly through DAF-16/FOXO. Humanin's lifespan extension also requires DAF-16. While these studies don't prove humanin acts through sirtuins, they establish that both operate through the same downstream effector, suggesting potential synergy or convergence.
Qin et al. (2018) demonstrated that chronic HNG treatment (4 mg/kg twice weekly for 14 months) prevented age-related myocardial fibrosis in mice, reducing collagen deposition, fibroblast proliferation, and apoptosis through upregulation of the Akt/GSK-3beta pathway.[13] SIRT1 also activates the Akt pathway and suppresses cardiac fibrosis in aging models, representing another point of convergence between humanin signaling and sirtuin function.
Peptide-Based Sirtuin Activation
Most sirtuin activators are small molecules. Resveratrol was the first widely studied SIRT1 activator (though its mechanism remains debated). SRT1720 and SRT2104 were designed as more specific SIRT1 activators by Sirtris Pharmaceuticals. Peptide-based activators represent a different approach.
Kumar et al. (2017) designed a tripeptide, CWR (cysteine-tryptophan-arginine), through molecular docking against the SIRT1 crystal structure. This three-amino-acid peptide activated SIRT1 through an allosteric mechanism, lowering the Michaelis constant (Km) rather than competing at the active site. The CWR peptide enhanced SIRT1 activity in purified enzyme, increased SIRT1 activity in serum from Alzheimer's disease patients, decreased p53 acetylation in neuroblastoma cells, and protected cells from amyloid-beta-induced death.[14]
This is a proof-of-concept study with no animal or human data, and the peptide's stability, bioavailability, and blood-brain barrier penetration are unknown. But it demonstrates a principle: even very short peptides can allosterically modulate sirtuin activity. Whether this translates beyond a cell culture model remains an open question.
GLP-1 Receptor Agonists and Sirtuin Signaling
An unexpected connection has emerged between GLP-1 receptor agonists (semaglutide, liraglutide, tirzepatide) and sirtuin pathways. Multiple studies now show these peptide drugs activate SIRT1 and SIRT3 as part of their downstream signaling.
Liraglutide attenuated high glucose-induced endothelial cell senescence through SIRT1-mediated deacetylation of p53 and p65 via the LARP7/SIRT1 pathway, directly linking GLP-1 receptor activation to sirtuin-dependent cellular rejuvenation. Abdelfattah et al. (2025) demonstrated that GLP-1 agonists reduced vascular calcification in aged rats through miR-34a/SIRT6/NRF2/HO-1 signaling.[4] Semaglutide protected against diabetes-associated cardiac inflammation via SIRT3-dependent activation of the RKIP pathway.[2]
These findings don't mean GLP-1 drugs are "sirtuin activators" in the way resveratrol was framed. Rather, sirtuin activation appears to be one downstream consequence of GLP-1 receptor signaling, likely mediated through AMPK and changes in cellular NAD+ metabolism. The metabolic improvements these drugs produce (weight loss, improved insulin sensitivity, reduced inflammation) create conditions favorable for sirtuin activity.
The Caloric Restriction Parallel
The GLP-1/sirtuin connection becomes more interesting when considered alongside caloric restriction. GLP-1 agonists reduce food intake and body weight, producing a state that partially mimics caloric restriction. Caloric restriction is the most robust activator of sirtuins in mammals. Jeromson et al. (2025) showed that semaglutide impacted skeletal muscle to a similar extent as caloric restriction in diet-induced obese mice, affecting the same mitochondrial and metabolic pathways. If GLP-1 agonists activate sirtuins, it may be partly because they reproduce the metabolic conditions (reduced caloric intake, improved insulin sensitivity, lower inflammation) that naturally drive sirtuin activity. The question is whether GLP-1 receptor signaling adds sirtuin activation beyond what weight loss and metabolic improvement alone would produce. Current evidence cannot separate these effects.
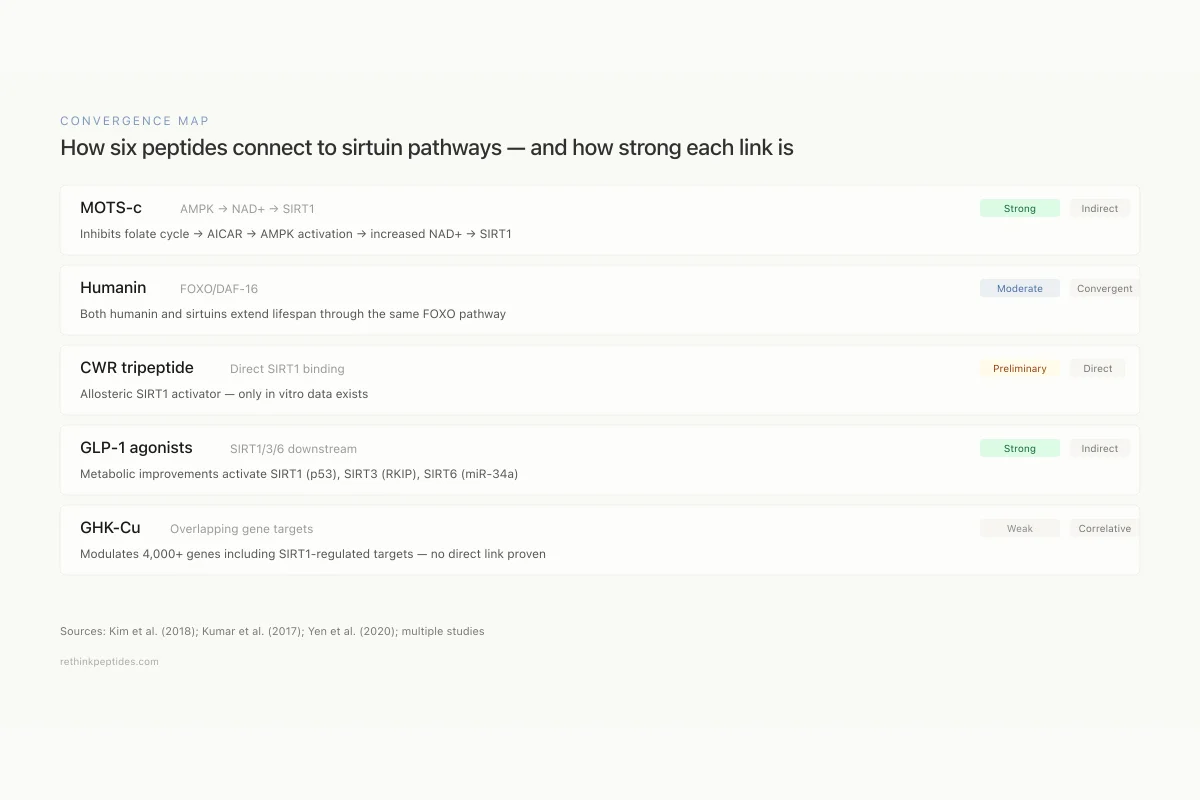
Convergence Map
How six peptides connect to sirtuin pathways — and how strong each link is
MOTS-c
AMPK → NAD+ → SIRT1
Inhibits folate cycle → AICAR → AMPK activation → increased NAD+/NADH → SIRT1
Humanin
FOXO/DAF-16
Both humanin and sirtuins extend lifespan through the same FOXO pathway
CWR tripeptide
Direct SIRT1 binding
Allosteric SIRT1 activator — only in vitro data exists
GLP-1 agonists
SIRT1/3/6 downstream
Metabolic improvements activate SIRT1 (p53), SIRT3 (RKIP), SIRT6 (miR-34a)
GHK-Cu
Overlapping gene targets
Modulates 4,000+ genes including SIRT1-regulated targets — no direct link proven
Cathelicidin-BF
AMPK/SIRT1/NF-κB
Reduces osteoarthritis inflammation through AMPK/SIRT1 in mice
Sources: Kim et al. (2018); Kumar et al. (2017); Yen et al. (2020); multiple studies
 View as image
View as imageOther Peptides in Sirtuin-Adjacent Pathways
GHK-Cu
The tripeptide GHK-Cu (glycyl-L-histidyl-L-lysine copper complex) modulates expression of over 4,000 human genes, including genes regulated by SIRT1. Pickart et al. (2012) demonstrated GHK-Cu's role in preventing oxidative stress and degenerative conditions of aging, with particular focus on cognitive health.[15] GHK-Cu upregulates antioxidant genes and suppresses inflammatory gene expression through mechanisms that overlap with SIRT1 transcriptional targets, though direct sirtuin modulation has not been established. For detailed GHK-Cu research, see GHK-Cu: The Copper Peptide That Modulates Over 4,000 Genes.
Epithalon
The tetrapeptide epithalon (Ala-Glu-Asp-Gly) induces telomerase activity and telomere elongation in human somatic cells.[16] SIRT6 is a known regulator of telomere maintenance, protecting telomeric chromatin through H3K9 deacetylation. Whether epithalon's telomerase activation involves SIRT6 remains untested, but both target the same cellular endpoint: telomere preservation. See Epithalon and Melatonin: The Pineal Gland Connection for more on epithalon research.
Cathelicidin-BF
Zhou et al. (2024) showed that cathelicidin-BF, an antimicrobial peptide, ameliorated osteoarthritis in mice through AMPK/SIRT1/NF-kappaB pathway regulation, directly demonstrating a naturally occurring peptide that modulates sirtuin signaling to reduce inflammation.[17]
FOXO-Activating Peptides
Du et al. (2025) demonstrated that ginseng oligopeptides extended lifespan in C. elegans through DAF-16/FOXO pathway activation, the same sirtuin-regulated longevity pathway.[18] While these oligopeptides don't directly activate sirtuins, they converge on the same transcriptional program that sirtuins regulate.
What the Evidence Does and Does Not Support
The peptide-sirtuin connection is real but nuanced. Here is what can be stated with confidence:
Supported by direct evidence:
- MOTS-c activates AMPK, which increases NAD+/NADH ratio and activates SIRT1
- The CWR tripeptide allosterically activates SIRT1 in vitro
- GLP-1 receptor agonists activate SIRT1, SIRT3, and SIRT6 as downstream effectors
- Cathelicidin-BF modulates inflammation through AMPK/SIRT1/NF-kappaB
- Humanin and sirtuins both regulate lifespan through FOXO/DAF-16
Not supported or unproven:
- No peptide has been shown to extend mammalian lifespan specifically through sirtuin activation
- Whether the sirtuin-longevity connection itself is causal in mammals remains actively debated
- Most peptide-sirtuin studies are in cell culture or animal models
- The CWR tripeptide has no in vivo data
- GHK-Cu and epithalon connections to sirtuins are correlative (shared pathway targets), not mechanistically established
The strongest case for peptide-sirtuin interaction comes from MOTS-c, where the mechanism (AMPK activation leading to SIRT1 activation) is well-characterized and the metabolic outcomes (improved insulin sensitivity, exercise-mimetic effects, stress resistance) align with known sirtuin functions.
The Convergence Problem in Longevity Research
One of the challenges in evaluating peptide-sirtuin connections is what might be called the convergence problem. AMPK, FOXO, NRF2, mTOR, and NF-kappaB are nodes in a densely interconnected signaling network. Almost any intervention that improves metabolic health will activate some of these pathways. Caloric restriction activates AMPK and SIRT1. Exercise activates AMPK and SIRT1. MOTS-c activates AMPK and (indirectly) SIRT1. GLP-1 agonists activate AMPK and SIRT1. The pattern is clear, but the specificity is not.
This convergence makes it difficult to determine whether a given peptide's longevity-relevant effects are mediated through sirtuins specifically, through parallel pathways, or through general metabolic improvement. The CWR tripeptide study is valuable precisely because it demonstrates direct, allosteric SIRT1 activation, bypassing the AMPK-NAD+ axis entirely. But one in vitro study doesn't resolve the broader question.
The practical implication is that peptide-sirtuin interactions should be understood as one layer of a multilayered system rather than as a simple cause-and-effect relationship. A peptide that activates AMPK will likely increase sirtuin activity, but it will also activate FOXO, inhibit mTOR, reduce NF-kappaB signaling, and improve mitochondrial function through multiple parallel mechanisms. Attributing the outcome to "sirtuin activation" alone would be an oversimplification.
Species Translation Concerns
The species translation problem also applies. Most sirtuin-longevity studies come from yeast, worms, and flies, organisms with one or two sirtuin genes compared to mammals' seven. C. elegans lifespan experiments, including the humanin and ginseng oligopeptide studies cited here, involve organisms that live weeks, not decades. Mouse studies provide mammalian data but with lifespans of 2-3 years. Whether the peptide-sirtuin interactions observed in these short-lived organisms predict effects in humans, who must maintain these systems across 70-90 years of NAD+ decline, oxidative damage, and genomic instability, is an unanswered question.
The most direct human evidence comes from observational data: humanin levels are higher in children of centenarians, MOTS-c levels decline with age and in metabolic disease, and GLP-1 agonists produce sirtuin activation alongside their metabolic benefits. None of this proves that manipulating peptide-sirtuin interactions will extend human lifespan, but it identifies these pathways as worthy of continued investigation.
The Bottom Line
Sirtuins sit at the intersection of energy metabolism, stress response, and aging. Multiple peptides interact with sirtuin pathways: MOTS-c activates AMPK upstream of SIRT1, humanin regulates lifespan through the same FOXO pathway sirtuins control, GLP-1 agonists activate SIRT1/3/6 as downstream effectors, and even simple tripeptides can allosterically activate SIRT1 in vitro. The biological convergence is clear, but the translational relevance remains uncertain. No peptide has extended mammalian lifespan through proven sirtuin activation, and the sirtuin-longevity link itself is more complex than early studies suggested.
Sources & References
- 1RPEP-07506·Wan, Wei et al. (2023). “MOTS-c: The Mitochondrial Peptide That Links Exercise, Metabolism, and Aging.” Journal of translational medicine.Study breakdown →PubMed →↩
- 2RPEP-12168·Lin, Kaibin et al. (2025). “How Semaglutide Protects Diabetic Hearts: A Specific Anti-Inflammatory Pathway Identified.” British journal of pharmacology.Study breakdown →PubMed →↩
- 3RPEP-10257·Byrne, Nikole J et al. (2025). “Sirtuin 4 accelerates heart failure development by enhancing reactive oxygen species-mediated profibrotic transcriptional signaling..” Journal of molecular and cellular cardiology plus.Study breakdown →PubMed →↩
- 4RPEP-09735·Abdelfattah, Amira Mohammed et al. (2025). “GLP-1 Agonist Liraglutide Reduces Vascular Calcification in Both Diabetic and Non-Diabetic Rats.” European journal of pharmacology.Study breakdown →PubMed →↩
- 5RPEP-03003·Lee, Changhan et al. (2016). “MOTS-c: A novel mitochondrial-derived peptide regulating muscle and fat metabolism..” Free radical biology & medicine.Study breakdown →PubMed →↩
- 6RPEP-06635·Yoon, Tae Kwan et al. (2022). “Exercise, Mitohormesis, and Mitochondrial ORF of the 12S rRNA Type-C (MOTS-c)..” Diabetes & metabolism journal.Study breakdown →PubMed →↩
- 7RPEP-03753·Kim, Kyung Hwa et al. (2018). “The Mitochondrial-Encoded Peptide MOTS-c Translocates to the Nucleus to Regulate Nuclear Gene Expression in Response to Metabolic Stress..” Cell metabolism.Study breakdown →PubMed →↩
- 8RPEP-15235·Gudiksen, Anders et al. (2026). “MOTS-c Peptide Improves Muscle Energy Production Efficiency.” Free radical biology & medicine.Study breakdown →PubMed →↩
- 9RPEP-05345·Dieli-Conwright, Christina M et al. (2021). “Exercise Boosts the Mitochondrial Peptide MOTS-c in Breast Cancer Survivors.” Scientific reports.Study breakdown →PubMed →↩
- 10RPEP-12342·Luo, Zhuoding et al. (2025). “People with Worse Sleep Apnea Have Lower Levels of the Mitochondrial Peptide MOTS-c.” Sleep and biological rhythms.Study breakdown →PubMed →↩
- 11RPEP-05924·Zempo, Hirofumi et al. (2021). “A pro-diabetogenic mtDNA polymorphism in the mitochondrial-derived peptide, MOTS-c..” Aging.Study breakdown →PubMed →↩
- 12RPEP-05221·Yen, Kelvin et al. (2020). “Mitochondrial Peptide Humanin Extends Lifespan and Improves Healthspan Across Multiple Species.” Aging.Study breakdown →PubMed →↩
- 13RPEP-03855·Qin, Qing et al. (2018). “Chronic treatment with the mitochondrial peptide humanin prevents age-related myocardial fibrosis in mice..” American journal of physiology. Heart and circulatory physiology.Study breakdown →PubMed →↩
- 14RPEP-03352·Kumar, Rahul et al. (2017). “A Three-Amino-Acid Peptide Activates the Longevity Enzyme SIRT1 and Protects Brain Cells From Alzheimer's Damage.” European journal of medicinal chemistry.Study breakdown →PubMed →↩
- 15RPEP-02037·Pickart, Loren et al. (2012). “The human tripeptide GHK-Cu in prevention of oxidative stress and degenerative conditions of aging: implications for cognitive health..” Oxidative medicine and cellular longevity.Study breakdown →PubMed →↩
- 16RPEP-00833·Khavinson, V Kh et al. (2003). “Epithalon Peptide Activates Telomerase and Lengthens Telomeres in Human Cells.” Bulletin of experimental biology and medicine.Study breakdown →PubMed →↩
- 17RPEP-09680·Zhou, Hao et al. (2024). “Cathelicidin Peptide Treats Osteoarthritis by Activating AMPK and Reducing Inflammation.” International immunopharmacology.Study breakdown →PubMed →↩
- 18RPEP-10777·Du, Qian et al. (2025). “Ginseng Oligopeptides Promote Longevity and Enhance Stress Resistance in Caenorhabditis elegans via the DAF-16/FOXO Pathway..” Antioxidants (Basel.Study breakdown →PubMed →↩