Humanin: The Cytoprotective Peptide from Your Mitochondria
Mitochondrial-Derived Peptides
24 amino acids
In 2001, researchers screening Alzheimer's brain tissue discovered a tiny peptide encoded in mitochondrial DNA that protected neurons from dying. They named it humanin.
Hashimoto et al., PNAS, 2001
Hashimoto et al., PNAS, 2001
If you only read one thing
Humanin is a tiny peptide that your mitochondria make, and it seems to protect cells from dying under stress. People who live past 100 tend to have more of it in their blood, and it declines as you age. In animal studies, it protects the brain, heart, and metabolism. The problem: no one has tested it in humans yet, and because it keeps cells alive, it might also keep cancer cells alive. It's one of the most fascinating peptides in aging research — but it's entirely pre-clinical.
The humanin peptide was discovered by accident. A team at Keio University in Tokyo was screening a cDNA library from the brain of an Alzheimer's patient, searching for genes that could rescue neurons from amyloid-beta toxicity. They found a short open reading frame in the mitochondrial genome that encoded a 24-amino acid peptide capable of preventing neuronal death across multiple Alzheimer's disease models. Since that 2001 discovery, humanin has emerged as the founding member of a new class of bioactive molecules: mitochondrial-derived peptides (MDPs).
Two decades of research have connected humanin to neuroprotection, cardiovascular protection, metabolic regulation, and aging. Circulating levels decline with age in mice and humans, yet remain elevated in centenarian offspring and in the naked mole-rat, a species that shows negligible senescence.[1] These observations have driven growing interest in humanin as both a biomarker and a potential therapeutic target. But the evidence is overwhelmingly preclinical, and a concerning connection to cancer progression complicates the picture.
Key Takeaways
- Humanin was discovered by accident in 2001 while scientists hunted for genes that protect brain cells in Alzheimer's.
- Your mitochondria don't just make energy — they also broadcast this tiny protective peptide throughout your body.
- People who live past 100 carry more humanin in their blood; levels drop as everyone else ages.
- In two decades of research, humanin has never been tested in humans — every finding is from cells or animals.
- A 14-month treatment in old mice reversed heart scarring and cut cardiac cell death.
- The peptide is so ancient that worms, mole-rats, and humans all carry nearly identical copies.
- Because humanin keeps stressed cells alive, it could also help cancer cells survive — that's the big unresolved concern.
What Is Humanin?
Humanin is a small peptide encoded by a short open reading frame within the MT-RNR2 gene, the 16S ribosomal RNA gene in the mitochondrial genome.[2] When produced inside mitochondria, it is 21 amino acids long. When produced in the cytosol using nuclear copies of the mitochondrial sequence, it extends to 24 amino acids. Both forms are biologically active. Its amino acid sequence (MAPRGFSCLLLLTSEIDLPVKRRA in the 24-residue form) contains no disulfide bonds and is remarkably resistant to aggregation, properties that distinguish it from many other small peptides.
This matters because most known peptide hormones are encoded in nuclear DNA. Humanin was the first peptide shown to be encoded in mitochondrial DNA and to function as a signaling molecule, fundamentally expanding what mitochondria were understood to do. Before humanin's discovery, mitochondria were primarily viewed as energy-producing organelles. The identification of humanin suggested they also function as endocrine organs, broadcasting stress signals to the rest of the cell and to distant tissues.[3]
Humanin circulates in human plasma at detectable levels, is present in cerebrospinal fluid, and has been identified in multiple tissue types including brain, skeletal muscle, heart, liver, kidney, and testes. The peptide is secreted from cells and can act in both autocrine (on the producing cell) and paracrine (on neighboring cells) fashion, as well as in an endocrine manner through systemic circulation.
Humanin is remarkably conserved across species. The peptide sequence is found in organisms ranging from nematodes to naked mole-rats to humans, suggesting an evolutionary origin spanning over 500 million years.[1] This degree of conservation typically indicates strong selective pressure to maintain the peptide's function.
How Humanin Works: Signaling Pathways
Humanin acts through multiple signaling pathways, which partly explains its broad range of protective effects.
Bax inhibition. One of humanin's best-characterized mechanisms is direct binding to Bax, a pro-apoptotic protein in the BCL-2 family. Humanin prevents Bax from translocating from the cytosol to the mitochondrial membrane, blocking a critical step in the apoptotic cascade. Reducing humanin expression with small interfering RNAs sensitizes cells to Bax-mediated death, confirming that endogenous humanin levels contribute to cell survival under stress.
Receptor signaling. Humanin binds the formylpeptide receptor-like 1 (FPRL-1), a G protein-coupled receptor, and the ciliary neurotrophic factor receptor (CNTFR)/WSX-1/gp130 trimeric receptor complex, activating downstream survival pathways. Through these receptor interactions, humanin activates the JAK2/STAT3 pathway, the ERK1/2 cascade, and the PI3K/AKT pathway.[4] These are canonical survival pathways that protect cells from oxidative stress, inflammation, and programmed death.
IGFBP-3 interaction. Humanin also binds insulin-like growth factor binding protein 3 (IGFBP-3) and antagonizes its pro-apoptotic activity. This interaction connects humanin to the insulin/IGF-1 signaling axis, one of the most studied longevity pathways in biology.[1]
AMPK activation. Recent work shows humanin-G (a synthetic analog) activates AMP-activated protein kinase (AMPK), particularly the alpha-1 catalytic subunit. In a hemorrhagic shock mouse model, humanin-G ameliorated lung injury through both AMPK-dependent mechanisms (reducing neutrophil infiltration) and AMPK-independent mechanisms (preserving alveolar structure).[6]
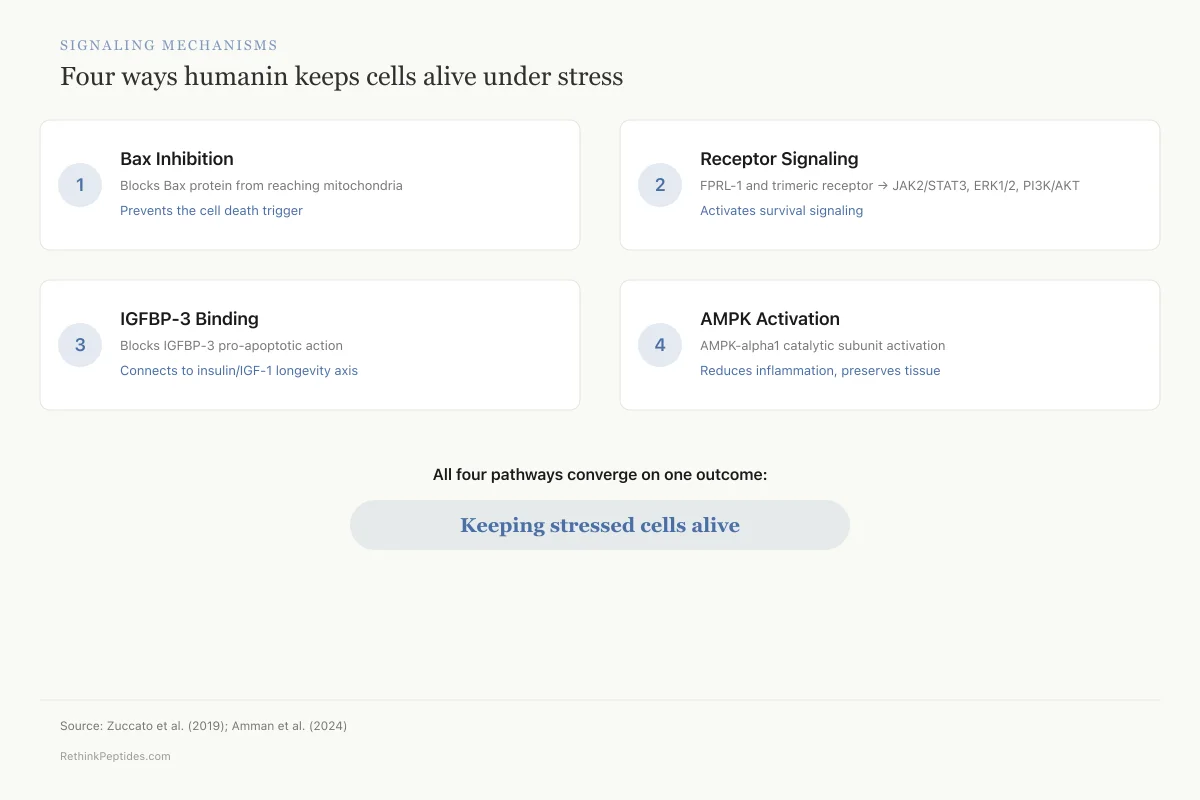
Signaling Mechanisms
Four ways humanin keeps cells alive under stress
Bax Inhibition
Blocks Bax → mitochondria translocation
Prevents the cell death trigger
FPRL-1 / Trimeric Receptor
JAK2/STAT3, ERK1/2, PI3K/AKT
Activates survival signaling
IGFBP-3 Binding
Blocks IGFBP-3 pro-apoptotic action
Connects to insulin/IGF-1 longevity axis
AMPK Activation
AMPK-alpha1 catalytic subunit
Reduces inflammation, preserves tissue
The pattern: All four pathways converge on the same outcome — preventing cells from dying when they're under stress. This multi-pathway protection is why humanin works across so many different tissues.
Source: Zuccato et al. (2019); Amman et al. (2024)
 View as image
View as imageHumanin and Neuroprotection
Neuroprotection is where humanin research began, and where the evidence is deepest.
The original discovery showed humanin rescued neurons from toxicity induced by multiple familial Alzheimer's disease genes and by amyloid-beta peptide. Subsequent work confirmed that humanin protects against a range of neurotoxic insults: serum deprivation, oxidative stress, and excitotoxicity.[5] The peptide has been detected in astrocytes, where it appears to prevent synapse loss in hippocampal neurons.
In the context of amyloid-beta, humanin shows a specific interaction. A 2024 study created a fusion peptide (HNSS) by merging SS-31 and a humanin analog (HNG). This hybrid showed 2-fold improved brain distribution over HNG alone, efficiently alleviated mitochondrial dysfunction through combined ROS scavenging and STAT3 activation, and reduced amyloid-beta oligomerization. In triple-transgenic Alzheimer's mice, HNSS treatment inhibited brain neuron loss and improved cognitive performance.[7]
Circulating humanin levels are reduced in patients with Alzheimer's disease and in those with MELAS (Mitochondrial Encephalopathy, Lactic Acidosis, and Stroke-like episodes).[1] Whether low humanin levels contribute to disease progression or simply reflect mitochondrial dysfunction remains an open question.
The neuroprotective evidence also extends beyond Alzheimer's. In Parkinson's disease models, humanin has shown protective effects against dopaminergic neuron loss. In stroke models, the peptide reduced infarct volume and improved functional recovery. These effects are consistent with humanin's general anti-apoptotic mechanism rather than any disease-specific action, which raises an important question: is humanin genuinely neuroprotective, or does it simply delay the death of cells that are already committed to dying? The distinction matters for therapeutic development. A peptide that genuinely prevents the cellular damage underlying neurodegeneration would have different clinical utility than one that merely prolongs cell survival without addressing the root cause.
All neuroprotection data comes from cell culture and animal models. No clinical trials have tested humanin or its analogs in patients with neurodegenerative disease.
Humanin and Cardiovascular Protection
Cardiac fibrosis increases with age and drives myocardial dysfunction. Qin et al. (2018) tested whether chronic humanin supplementation could prevent this process. Female mice at 18 months received intraperitoneal injections of the humanin analog HNG (4 mg/kg, twice weekly) for 14 months and were euthanized at 32 months. HNG treatment increased the ratio of cardiomyocytes to fibroblasts, reduced collagen deposition, inhibited fibroblast proliferation, and attenuated expression of TGF-beta1, FGF-2, and MMP-2. Myocardial apoptosis was also reduced. The mechanism involved upregulation of the Akt/GSK-3beta pathway.[8]
This was the first demonstration that exogenous humanin treatment could attenuate age-related cardiac remodeling in a mammal. The 14-month treatment duration and the assessment at 32 months (extreme old age for mice) make this study particularly noteworthy for its long-term design.
Beyond fibrosis, humanin has shown protective effects in models of atherosclerosis and myocardial ischemia-reperfusion injury. The review by Zuccato et al. (2019) catalogued humanin's cardiovascular protective actions across multiple model systems, noting consistent anti-apoptotic and anti-inflammatory effects in cardiac tissue.[5]
The cardiovascular data, like the neuroprotection data, is entirely preclinical. No human cardiovascular trials exist.
Humanin, Metabolism, and Insulin Sensitivity
Humanin interacts with the insulin/IGF-1 signaling axis at multiple levels. Treating middle-aged mice with HNG twice weekly improved metabolic healthspan parameters and reduced inflammatory markers.[1] Humanin appears to function as a central regulator of peripheral insulin action, improving glucose handling and insulin sensitivity in rodent models.
The broader MDP family shows consistent metabolic effects. Merry et al. (2020) reviewed evidence that metabolic conditions like obesity, diabetes, and aging are associated with lower circulating MDP levels. In contrast, muscle MDP expression is upregulated in response to mitochondrial stress, including exercise, certain mtDNA mutations, and healthy aging. This suggests a tissue-specific compensatory response aimed at restoring cellular homeostasis.[3]
Treatment of rodents with humanin, MOTS-c, and SHLP2 enhanced insulin sensitivity and offered protection against age-associated metabolic disorders. Whether these effects translate to humans at therapeutic doses is unknown. The relationship between humanin and insulin sensitivity is bidirectional: humanin improves insulin action, and insulin signaling appears to regulate humanin expression. This feedback loop suggests that metabolic disease and humanin deficiency may reinforce each other in a downward spiral.
In type 2 diabetes models, humanin administration improved glucose tolerance and reduced fasting insulin levels, consistent with improved insulin sensitivity rather than increased insulin secretion. The peptide's interaction with IGFBP-3, which normally promotes apoptosis of pancreatic beta cells, may also provide indirect protection to insulin-producing cells under metabolic stress.
Thoudam et al. (2026) extended this metabolic story to the liver, reviewing evidence that MDPs regulate hepatic metabolism and may serve as therapeutic targets for liver disease.[9] Given the liver's central role in glucose and lipid metabolism, this represents a substantial expansion of the MDP landscape. Humanin's effects on hepatic lipid accumulation and mitochondrial fatty acid oxidation suggest it may influence nonalcoholic fatty liver disease progression, though this remains early-stage research.
Humanin Levels Decline with Age
One of the most compelling observations about humanin is its age-dependent decline.
Yen et al. (2020) measured circulating humanin levels across species and found consistent age-related decreases in mice and humans. In C. elegans, humanin overexpression was sufficient to extend lifespan through a mechanism dependent on daf-16/FOXO, a transcription factor central to longevity pathways across species.[1]
Three observations from the Yen study are particularly striking:
- Centenarian offspring: Children of centenarians, who are statistically more likely to become centenarians themselves, had higher circulating humanin levels than age-matched controls.
- Naked mole-rats: Humanin levels remained stable across the lifespan in naked mole-rats, a species that exhibits negligible senescence and lives roughly 30 years (about 10 times the lifespan of similarly sized rodents).
- Disease states: Humanin levels were decreased in Alzheimer's disease and MELAS patients.
These correlations do not prove causation. Higher humanin levels in centenarian offspring could reflect better mitochondrial function rather than a direct protective effect. But the convergent evidence across species and disease states is difficult to dismiss.
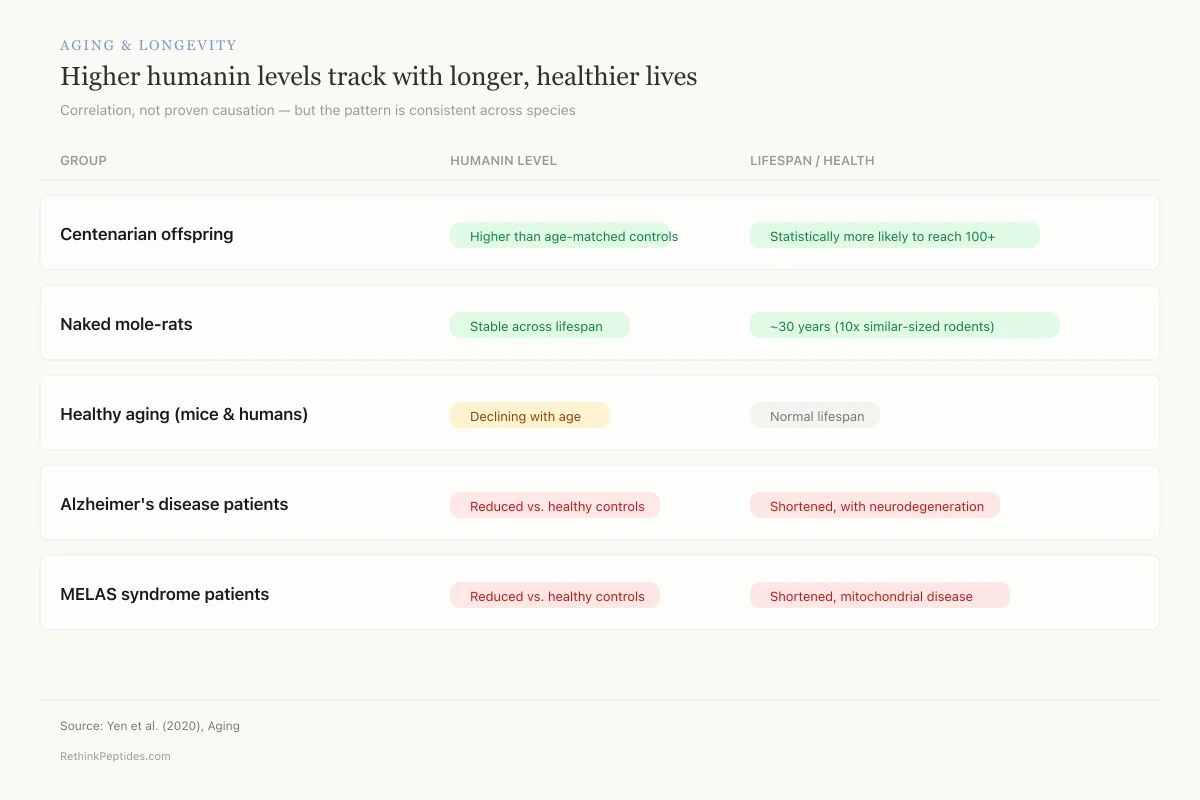
Aging & Longevity
Higher humanin levels track with longer, healthier lives
Correlation, not proven causation — but the pattern is consistent across species
Centenarian offspring
Higher than age-matched controlsStatistically more likely to reach 100+Naked mole-rats
Stable across lifespan~30 years (10× similar-sized rodents)Healthy aging (mice & humans)
Declining with ageNormal lifespanAlzheimer's disease patients
Reduced vs. healthy controlsShortened, with neurodegenerationMELAS syndrome patients
Reduced vs. healthy controlsShortened, mitochondrial diseaseSource: Yen et al. (2020), Aging
 View as image
View as imageExercise also modulates MDP levels. Von Walden et al. (2021) found that acute endurance exercise stimulated circulating levels of mitochondrial-derived peptides in healthy humans, providing evidence that physical activity engages the same retrograde mitochondrial signaling pathways that humanin operates through.[10]
The Broader MDP Family: Beyond Humanin
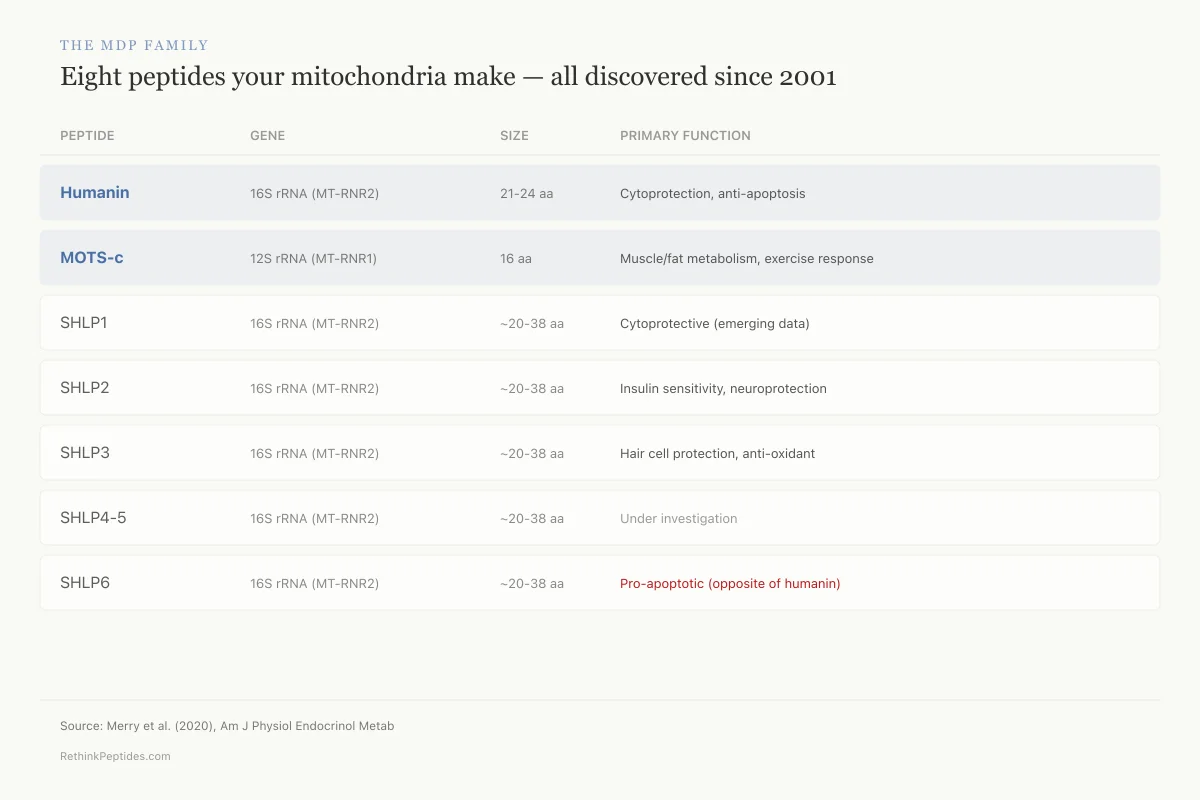
Humanin was the first identified MDP, but it is not alone. To date, eight MDPs have been identified: humanin, MOTS-c, and six small humanin-like peptides (SHLPs 1-6).[3]
Humanin and SHLPs 1-6 are all encoded in the 16S ribosomal RNA (MT-RNR2) gene. MOTS-c is encoded in the 12S ribosomal RNA (MT-RNR1) gene. Despite sharing a mitochondrial origin, these peptides appear to act through different pathways and affect different tissues.
MOTS-c regulates muscle and fat metabolism, with particular relevance to exercise physiology. Lee et al. (2016) characterized MOTS-c as a regulator of skeletal muscle metabolism that targets the folate-methionine cycle.[11] Wan et al. (2023) provided a comprehensive review of MOTS-c's effects on stress resistance, metabolism, and aging, highlighting overlapping but distinct functions compared to humanin.[12]
SHLPs have received less attention, but emerging data shows cytoprotective effects. Lu et al. (2024) demonstrated that both HNG (a humanin analog) and SHLP3 protected cochlear hair cells against gentamicin-induced toxicity, reducing oxidative stress and inflammatory gene expression through modulation of AKT and AMPK-alpha pathways.[13] This is notable because it shows HNG and SHLP3 acting through partly overlapping but likely distinct signaling mechanisms.
The MDP Family
Eight peptides your mitochondria make — all discovered since 2001
Humanin and MOTS-c are best studied; SHLPs are just emerging
Humanin
Cytoprotection, anti-apoptosis
MOTS-c
Muscle/fat metabolism, exercise response
SHLP1
Cytoprotective (emerging data)
SHLP2
Insulin sensitivity, neuroprotection
SHLP3
Hair cell protection, anti-oxidant
SHLP4
Under investigation
SHLP5
Under investigation
SHLP6
Pro-apoptotic (opposite of humanin)
Source: Merry et al. (2020), Am J Physiol Endocrinol Metab
 View as image
View as imageThe relationship between MDPs raises a fundamental question: are these peptides part of a coordinated retrograde signaling network that communicates mitochondrial status to the rest of the cell and to distal tissues? Or do they act independently? Current evidence favors the network hypothesis, but definitive proof requires more work.
For a deeper look at the mitochondria-targeted synthetic peptide SS-31 (elamipretide), which operates on mitochondrial membranes rather than being encoded by mitochondrial DNA, see the dedicated article.
Humanin Analogs: Engineering a More Potent Peptide
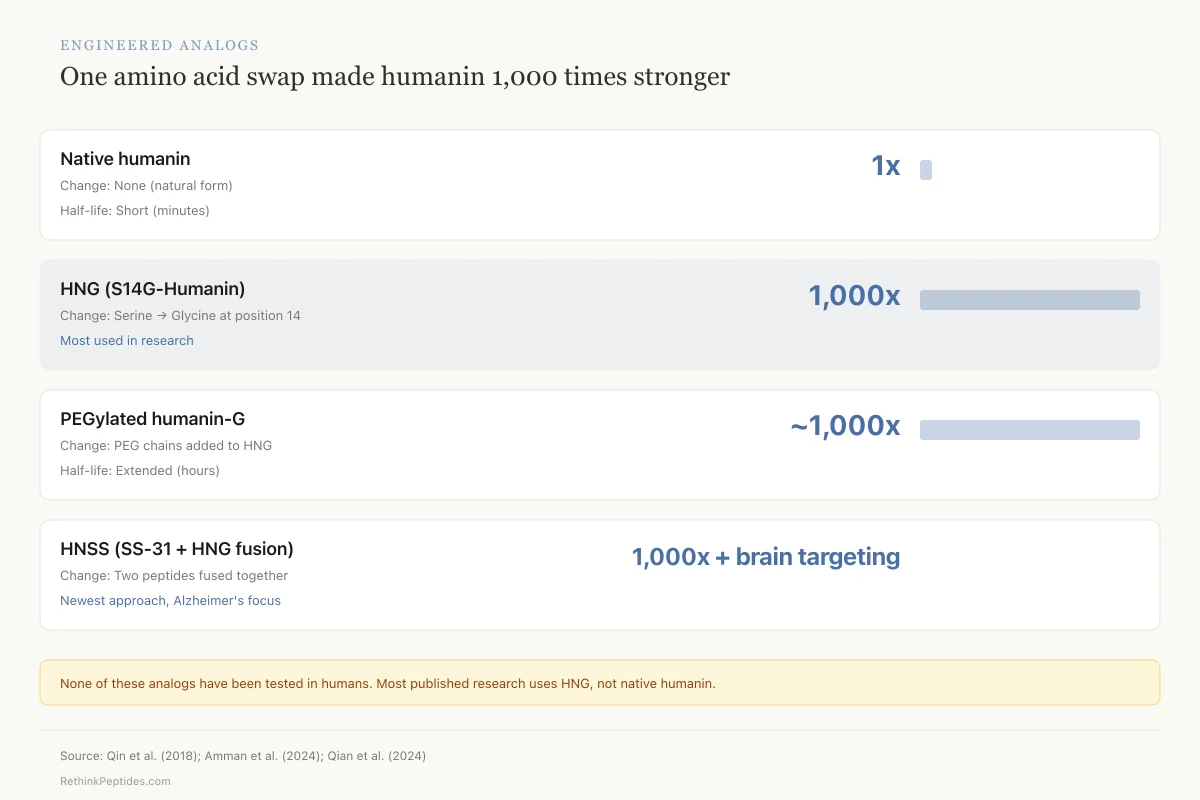
Natural humanin has a short half-life in circulation, limiting its therapeutic utility. This has driven the development of synthetic analogs with enhanced potency and stability.
HNG (S14G-Humanin): The single amino acid substitution of glycine for serine at position 14 produces a peptide approximately 1,000 times more potent than native humanin in cell-based assays. The mechanism for this dramatic potency increase is not fully understood, but the substitution appears to alter the peptide's conformation in a way that enhances receptor binding without changing its fundamental signaling profile. HNG is the most widely used analog in preclinical research and was the form used in the Qin et al. cardiac fibrosis study and the Lu et al. cochlear hair cell study.[8][13] Most published preclinical efficacy data uses HNG rather than native humanin, which means the therapeutic window and safety profile of native humanin remains less characterized.
PEGylated humanin-G: Adding polyethylene glycol chains extends circulating half-life. Amman et al. (2024) used this form in their hemorrhagic shock model and observed tissue protection in both AMPK-alpha1 wild-type and knockout mice, indicating the PEGylated form retains activity through multiple pathways.[6]
Fusion peptides: The HNSS hybrid (SS-31 fused with HNG) represents a newer strategy that combines two protective peptide motifs into a single molecule. The design mimics cell-penetrating peptide architecture to improve brain permeability, addressing one of the key barriers to CNS drug delivery.[7]
Engineered Analogs
One amino acid swap made humanin 1,000 times stronger
Key caveat: Most published humanin research uses HNG, not the natural peptide. We don't fully understand why swapping one amino acid produces 1,000× more potency. None of these analogs have been tested in humans.
Source: Qin et al. (2018); Amman et al. (2024); Qian et al. (2024)
 View as image
View as imageBeyond synthetic analogs, Zuccato et al. (2019) discussed gene therapy approaches: local administration of viral vectors that overexpress or silence endogenous humanin could offer targeted tissue-specific effects.[5] None of these approaches have reached human trials.
Humanin Beyond the Brain and Heart
Humanin's cytoprotective effects extend beyond the nervous and cardiovascular systems.
Reproductive biology. Humanin acts as a cytoprotective factor in ovarian granulosa cells. Downregulating humanin in the ovary modified histoarchitecture and increased apoptosis, while exogenous humanin treatment increased cell viability in a granulosa tumor cell line. This suggests the peptide is involved in regulating folliculogenesis and may connect mitochondrial function to fertility.[4]
Lung injury. Amman et al. (2024) showed humanin-G ameliorated hemorrhage-induced acute lung injury in mice, with sex-dependent characteristics. Male mice showed more pronounced neutrophil infiltration, and AMPK-alpha1 knockout males experienced greater hemodynamic instability. Humanin-G protected alveolar structure regardless of AMPK-alpha1 status, but its effect on neutrophil infiltration required AMPK-alpha1.[6]
Hearing protection. HNG and SHLP3 protected cochlear hair cells from gentamicin toxicity, a major cause of drug-induced hearing loss. The protection correlated with reduced oxidative stress and modulation of AKT and AMPK-alpha.[13]
This breadth of tissue protection is consistent with humanin's fundamental role in the cellular stress response rather than any tissue-specific function. Wherever mitochondria are under stress and cells are at risk of apoptosis, humanin appears to offer a protective signal.
The Cancer Complexity
Humanin's anti-apoptotic properties create a double-edged problem in cancer biology. The same mechanisms that protect healthy cells from death may also protect tumor cells from apoptosis.
Zuccato et al. (2019) flagged this directly: controversy exists regarding humanin's role in cancer progression and chemoresistance. Some studies show humanin promotes tumor survival and may interfere with chemotherapy-induced apoptosis.[5] A 2020 study in Scientific Reports found humanin promoted tumor progression in experimental triple-negative breast cancer models.
Safety
CriticalHumanin may help tumors survive
Concern
The same anti-death mechanism that makes humanin protective in healthy cells could also protect cancer cells. Studies in triple-negative breast cancer models found humanin promoted tumor progression, and the peptide may interfere with chemotherapy by blocking the cell death that chemo is trying to trigger.
What the research says
This is unresolved. Researchers have proposed tissue-targeted delivery (giving humanin only to the heart or brain, not systemically) to avoid feeding existing tumors. No humanin analog that selectively protects healthy cells but not cancer cells has been developed.
Particularly relevant for: Anyone with existing or undetected cancer, or elevated cancer risk
What to do
This concern must be resolved before any human trials can proceed. The Zuccato 2019 review explicitly called this the primary barrier to clinical translation.
Zuccato et al. (2019), Expert Opinion on Therapeutic Targets
This is not a minor caveat. Any therapeutic strategy that elevates systemic humanin levels could theoretically support existing tumors. Until the relationship between humanin and cancer biology is better characterized, this represents a genuine barrier to clinical translation.
The Zuccato review explicitly stated that "controversy on the role of HN in cancer progression and chemoresistance should be addressed before the translation of these therapeutic approaches."[5]
One potential resolution involves tissue-targeted delivery rather than systemic humanin elevation. If humanin could be administered locally to cardiac tissue after a heart attack, or directly to the brain in neurodegeneration, the cancer risk from systemic exposure might be avoided. The fusion peptide approach (HNSS) represents a step in this direction by engineering tissue selectivity into the peptide itself. Another approach is to develop humanin analogs that retain cytoprotective activity in normal cells but lack the ability to protect tumor cells, though no such analog has been reported.
Where the Evidence Stands
Humanin research spans over two decades and includes robust preclinical evidence of neuroprotection, cardioprotection, metabolic improvement, and lifespan extension. The correlative human data, particularly in centenarian offspring and across disease states, is compelling. But several realities shape how this evidence should be interpreted:
No human clinical trials. Humanin has not been tested in any human therapeutic trial. All efficacy data comes from cell culture, C. elegans, and rodent models. The leap from mouse to human is substantial, and many promising preclinical peptide therapies have failed in translation.
Cancer risk is unresolved. The anti-apoptotic mechanism that makes humanin protective in healthy cells may be harmful in the context of existing tumors. This must be resolved before any clinical application.
Biomarker potential is clearer than therapeutic potential. Circulating humanin levels correlate with aging, disease states, and longevity. As a diagnostic or prognostic biomarker, humanin may reach clinical utility sooner than as a therapeutic.
The MDP field is young. Eight peptides identified in 25 years is a small number, and the interactions between them are poorly understood. Humanin does not act in isolation. Its effects are likely modulated by MOTS-c, SHLPs, and other factors yet to be discovered.
Dosing and pharmacokinetics are uncharacterized in humans. The doses used in mouse studies (typically 4 mg/kg of HNG, twice weekly by intraperitoneal injection) cannot be directly translated to human equivalent doses without pharmacokinetic studies. Humanin's short half-life in circulation, the need for injection, and the absence of oral bioavailability data represent practical barriers alongside the biological ones.
Redundancy in the MDP system is unknown. If humanin is part of a network of mitochondrial signaling peptides, boosting one member of that network may have unpredictable effects on the others. The compensatory relationships between humanin, MOTS-c, and SHLPs are poorly mapped.
What humanin has accomplished is conceptual: it demonstrated that mitochondria encode signaling peptides that influence aging, metabolism, and cell survival across species. That idea alone has opened an entirely new branch of peptide biology.
The Bottom Line
Humanin is a 24-amino acid mitochondrial-derived peptide with consistent cytoprotective effects across cell types and organ systems in preclinical models. Circulating levels decline with age and disease, and are elevated in centenarian offspring, linking the peptide to human longevity. All therapeutic evidence remains preclinical, and an unresolved connection to tumor survival complicates any path to clinical use.
Sources & References
- 1RPEP-05221·Yen, Kelvin et al. (2020). “Mitochondrial Peptide Humanin Extends Lifespan and Improves Healthspan Across Multiple Species.” Aging.Study breakdown →PubMed →↩
- 2RPEP-04424·Popov, Lucia-Doina (2019). “Mitochondrial Peptides: From Humanin to MOTS-c and Targeted Drug Delivery.” Cell and tissue research.Study breakdown →PubMed →↩
- 3RPEP-04997·Merry, Troy L et al. (2020). “Mitochondrial-Derived Peptides: How Your Mitochondria Signal Metabolic Protection.” American journal of physiology. Endocrinology and metabolism.Study breakdown →PubMed →↩
- 4RPEP-04608·Zuccato, Camila Florencia et al. (2019). “Mitochondrial-derived peptide humanin as therapeutic target in cancer and degenerative diseases..” Expert opinion on therapeutic targets.Study breakdown →PubMed →↩
- 5RPEP-04608·Zuccato, Camila Florencia et al. (2019). “Mitochondrial-derived peptide humanin as therapeutic target in cancer and degenerative diseases..” Expert opinion on therapeutic targets.Study breakdown →PubMed →↩
- 6RPEP-07748·Amman, Allison M et al. (2024). “Humanin-G Protected Mouse Lungs After Hemorrhagic Shock Through Both AMPK-Dependent and Independent Pathways.” Biomedicines.Study breakdown →PubMed →↩
- 7RPEP-09102·Qian, Kang et al. (2024). “A Hybrid Peptide That Crosses Into the Brain to Fight Alzheimer's.” Asian journal of pharmaceutical sciences.Study breakdown →PubMed →↩
- 8RPEP-03855·Qin, Qing et al. (2018). “Chronic treatment with the mitochondrial peptide humanin prevents age-related myocardial fibrosis in mice..” American journal of physiology. Heart and circulatory physiology.Study breakdown →PubMed →↩
- 9RPEP-16248·Thoudam, Themis et al. (2026). “Peptides Made by Mitochondria Could Protect Against Fatty Liver Disease.” Hepatology communications.Study breakdown →PubMed →↩
- 10RPEP-05845·von Walden, Ferdinand et al. (2021). “Acute endurance exercise stimulates circulating levels of mitochondrial-derived peptides in humans..” Journal of applied physiology (Bethesda.Study breakdown →PubMed →↩
- 11RPEP-03003·Lee, Changhan et al. (2016). “MOTS-c: A novel mitochondrial-derived peptide regulating muscle and fat metabolism..” Free radical biology & medicine.Study breakdown →PubMed →↩
- 12RPEP-07506·Wan, Wei et al. (2023). “MOTS-c: The Mitochondrial Peptide That Links Exercise, Metabolism, and Aging.” Journal of translational medicine.Study breakdown →PubMed →↩
- 13RPEP-08772·Lu, Yu et al. (2024). “Mitochondrial-derived peptides, HNG and SHLP3, protect cochlear hair cells against gentamicin..” Cell death discovery.Study breakdown →PubMed →↩