MOTS-c: The Mitochondrial Exercise Peptide
MOTS-c and Mitochondrial Peptides
12-fold increase after exercise
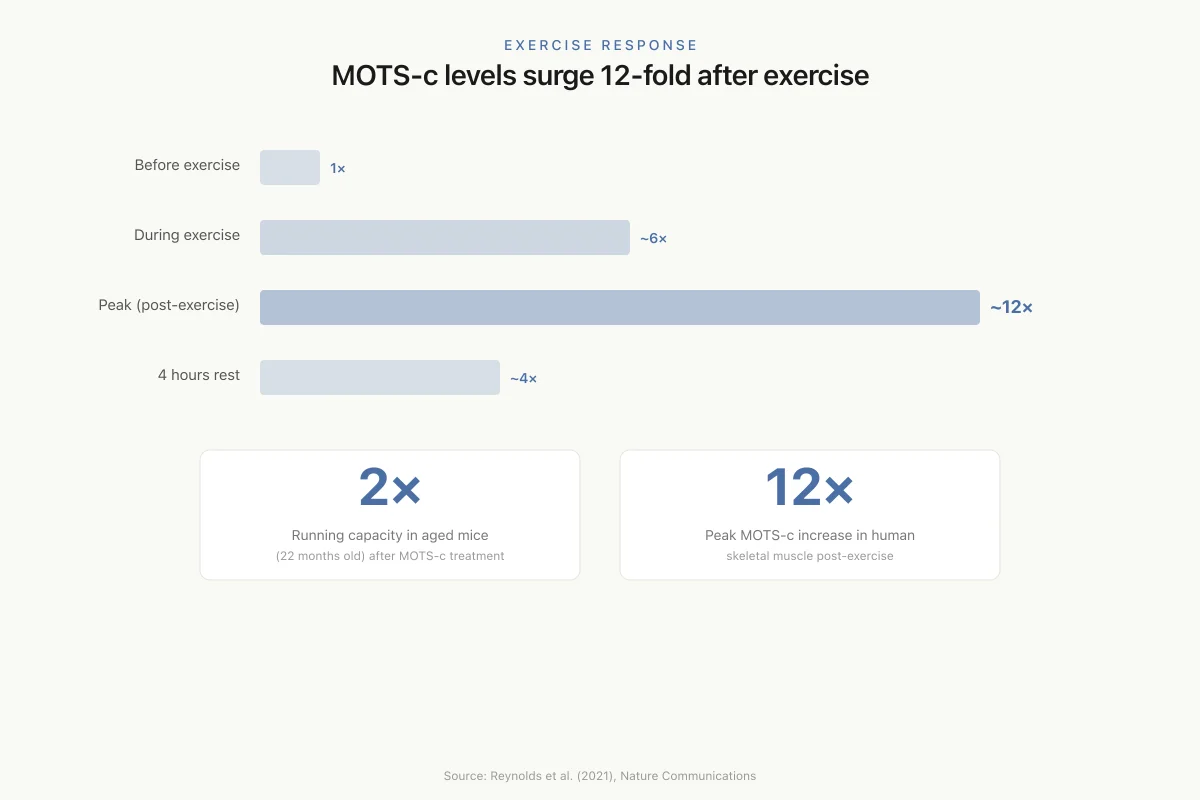
In human skeletal muscle, MOTS-c levels increased nearly 12-fold after exercise and remained partially elevated after a four-hour rest, linking this mitochondrial peptide directly to physical activity.
Reynolds et al., Nature Communications, 2021
Reynolds et al., Nature Communications, 2021
If you only read one thing
MOTS-c is a tiny peptide made by your mitochondria — the power plants inside your cells — that mimics some of the metabolic benefits of exercise. When you work out, MOTS-c levels in your muscles jump 12-fold. In old mice, injecting MOTS-c doubled their running capacity. It works by flipping the same metabolic switch (AMPK) that exercise activates, improving how your body handles sugar and burns fat. Your levels drop as you age and in diabetes. No human trials yet, but it's one of the most exciting peptides in metabolic research.
In 2015, Changhan Lee and colleagues at the USC Leonard Davis School of Gerontology published a discovery in Cell Metabolism that opened a new chapter in peptide biology. They identified a 16-amino-acid peptide encoded not by the nuclear genome but by the mitochondrial genome, specifically within the 12S ribosomal RNA gene (MT-RNR1). They named it MOTS-c (mitochondrial open reading frame of the 12S rRNA type-c) and showed that it regulated metabolic homeostasis: injecting MOTS-c into mice prevented both age-dependent and high-fat-diet-induced insulin resistance, reduced obesity, and enhanced glucose metabolism.[1]
The discovery was significant for two reasons. First, it demonstrated that mitochondria, long understood as energy-producing organelles, encode peptide hormones that regulate whole-body metabolism. Second, MOTS-c's effects resembled those of exercise: improved insulin sensitivity, enhanced glucose uptake, increased fat oxidation, and activation of the master metabolic sensor AMPK. Reynolds et al. (2021) later confirmed this connection in Nature Communications, showing that MOTS-c is directly induced by exercise in human skeletal muscle, with levels rising nearly 12-fold after physical activity.[2]
This article covers what MOTS-c is, how it works, what the animal and human data show, and where the limitations lie. For the AMPK mechanism in detail, see How MOTS-c Activates AMPK and Improves Insulin Sensitivity. For the age-related decline, see MOTS-c and Aging: Why Mitochondrial Peptides Decline with Age. For the exercise capacity data, see MOTS-c and Physical Performance: Can a Peptide Replace a Workout?. For another mitochondrial-derived peptide, see Humanin: The Cytoprotective Peptide from Your Mitochondria.
Key Takeaways
- MOTS-c is a peptide coded inside your mitochondria that behaves like a chemical version of exercise.
- When you work out, MOTS-c levels in your muscles jump roughly 12-fold.
- In old mice, a single peptide doubled running endurance — no training required.
- Your mitochondria aren't just batteries — they release signaling peptides that direct how your whole body uses energy.
- MOTS-c activates AMPK, the same metabolic switch that exercise and metformin hit.
- No human trials have tested MOTS-c directly yet — all the headline results come from mice.
- Your MOTS-c levels fall with age and are lower in people with type 2 diabetes.
What MOTS-c is: a peptide from the mitochondrial genome
Mitochondria carry their own small circular genome (mtDNA), approximately 16,500 base pairs encoding 37 genes: 13 proteins of the electron transport chain, 22 transfer RNAs, and 2 ribosomal RNAs. For decades, these 37 genes were considered the complete mitochondrial output. MOTS-c was one of the first discoveries to change that understanding.
MOTS-c is encoded within the 12S ribosomal RNA gene (MT-RNR1), not as a traditional open reading frame but as a short ORF embedded within the rRNA sequence. Its amino acid sequence is MRWQEMGYIFYPRKLR (16 residues). It is translated on mitochondrial ribosomes but then exported from the mitochondria into the cytoplasm and, under metabolic stress, translocated to the nucleus where it regulates gene expression.[1]
This mitochondria-to-nucleus signaling is called retrograde signaling, and it represents a fundamental revision of the traditional view of mitochondria as passive energy producers under nuclear control. MOTS-c is part of a growing family of mitochondrial-derived peptides (MDPs) that includes humanin (discovered in 2001) and SHLP1-6 (small humanin-like peptides). Together, these MDPs suggest that mitochondria function as signaling organelles that communicate their metabolic state to the rest of the cell and, through secretion into the bloodstream, to the entire organism.
Lee et al. (2016) published a follow-up characterizing MOTS-c as a regulator of both muscle and fat metabolism, showing that the peptide's effects were not limited to glucose handling but included modulation of lipid metabolism and body composition.[3]
The concept of mitochondrial-derived peptides challenges a century-old assumption about mitochondrial biology. The endosymbiotic origin of mitochondria (they were once free-living bacteria engulfed by ancestral eukaryotic cells approximately 2 billion years ago) means their genome is a relic of their independent past. The discovery that this ancient genome encodes bioactive signaling peptides suggests that the host-mitochondria relationship is more interactive than the master-servant model that dominated cell biology. Mitochondria are not just following nuclear orders; they are reporting back on their metabolic state through peptide signals that influence nuclear gene expression, systemic metabolism, and whole-organism physiology.
The AMPK mechanism: how MOTS-c mimics exercise
MOTS-c's metabolic effects converge on AMPK (AMP-activated protein kinase), the cell's master energy sensor. AMPK is activated when cellular energy is depleted (high AMP-to-ATP ratio), as occurs during exercise. Once active, AMPK triggers a cascade of metabolic adaptations: increased glucose uptake, enhanced fatty acid oxidation, mitochondrial biogenesis, and inhibition of energy-consuming processes like protein synthesis and lipogenesis.
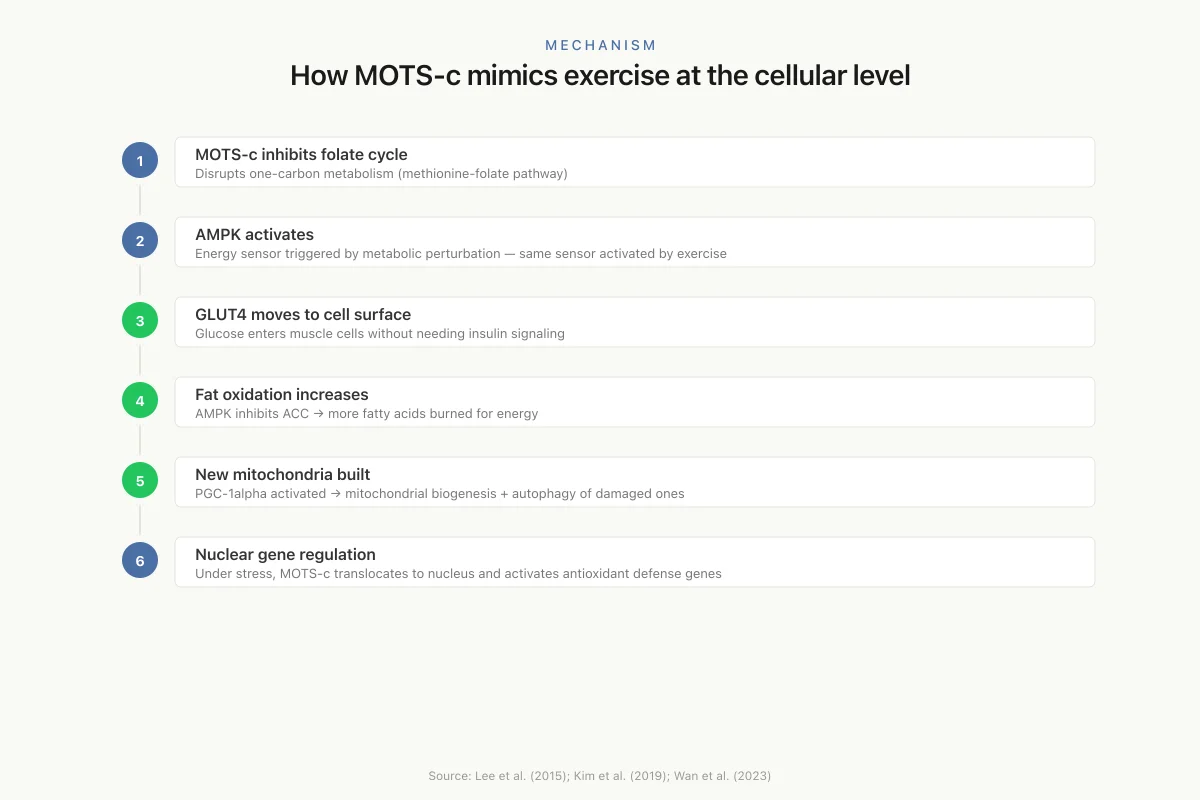
MOTS-c activates AMPK through an unusual upstream mechanism. Rather than directly binding AMPK or altering the AMP/ATP ratio, MOTS-c inhibits the folate-methionine cycle (also called one-carbon metabolism). The folate cycle is essential for nucleotide synthesis and methylation reactions. By partially inhibiting this cycle, MOTS-c creates a metabolic perturbation that triggers AMPK activation as a compensatory response.[1]
Kim et al. (2019) provided detailed metabolomic evidence for this mechanism, showing that MOTS-c treatment in mice altered the methionine-folate-purine pathway and that the downstream metabolic effects (enhanced insulin sensitivity, altered plasma metabolite profiles) were consistent with AMPK-mediated metabolic reprogramming.[4]
The AMPK activation drives GLUT4 translocation to the cell surface, increasing glucose uptake into skeletal muscle. This is the same mechanism by which exercise improves insulin sensitivity: muscle contraction activates AMPK, which drives GLUT4 to the membrane, allowing glucose to enter the cell without requiring insulin signaling. MOTS-c achieves a pharmacologically similar outcome through a different initiating stimulus.
AMPK also promotes mitochondrial biogenesis through PGC-1alpha activation, enhances fatty acid oxidation by phosphorylating and inhibiting acetyl-CoA carboxylase, and activates autophagy (including mitophagy, the selective degradation of damaged mitochondria). These downstream effects collectively improve cellular energy efficiency: more mitochondria, better mitochondria, more fat burning, and less reliance on glucose as a fuel source. For the full mechanistic details, see How MOTS-c Activates AMPK and Improves Insulin Sensitivity.
Under metabolic stress, MOTS-c translocates from the cytoplasm to the nucleus in an AMPK-dependent manner, where it binds to antioxidant response elements (AREs) and regulates stress-responsive gene expression. This nuclear translocation represents a direct signaling pathway from mitochondria to the nuclear genome, allowing the cell's energy-producing organelles to influence gene transcription based on metabolic conditions. The genes regulated through this pathway include those involved in antioxidant defense, cellular stress resistance, and metabolic adaptation, creating a coordinated response to metabolic challenges that is orchestrated from the mitochondria rather than from nuclear command.[5]
Mechanism
How MOTS-c mimics exercise at the cellular level
MOTS-c inhibits folate cycle
Disrupts one-carbon metabolism (methionine-folate pathway)
AMPK activates
Energy sensor triggered by metabolic perturbation — same sensor activated by exercise
GLUT4 moves to cell surface
Glucose enters muscle cells without needing insulin signaling
Fat oxidation increases
AMPK inhibits ACC → more fatty acids burned for energy
New mitochondria built
PGC-1alpha activated → mitochondrial biogenesis + autophagy of damaged ones
Nuclear gene regulation
Under stress, MOTS-c translocates to nucleus and activates antioxidant defense genes
Source: Lee et al. (2015); Kim et al. (2019); Wan et al. (2023)
 View as image
View as imageThe fact that MOTS-c operates through folate cycle inhibition rather than direct AMPK binding has pharmacological implications. Metformin, the most widely prescribed diabetes drug, also activates AMPK (though through a different upstream mechanism involving complex I inhibition). Whether MOTS-c and metformin would produce additive, synergistic, or redundant effects on AMPK-dependent metabolism has not been tested but represents an obvious question for combination therapy research.
The exercise connection
Reynolds et al. (2021) published the study that earned MOTS-c its "exercise mimetic" label. In human subjects, exercise induced a nearly 12-fold increase in MOTS-c levels in skeletal muscle, with significant elevation also detected in circulating blood. The levels remained partially elevated after a four-hour rest period, suggesting sustained post-exercise MOTS-c signaling.[2]
Exercise Response
MOTS-c levels in skeletal muscle surge 12-fold after exercise
2×
Running capacity in aged mice (22 months) after MOTS-c treatment
12×
Peak MOTS-c increase in human skeletal muscle post-exercise
Source: Reynolds et al. (2021), Nature Communications
 View as image
View as imageIn the mouse component of the study, MOTS-c treatment significantly enhanced physical performance across all age groups: young (2 months), middle-aged (12 months), and old (22 months). The most dramatic result was in old mice, where MOTS-c treatment doubled treadmill running capacity. The improvement was associated with enhanced mitochondrial function, improved muscle homeostasis, and activation of exercise-responsive gene networks in skeletal muscle.[2]
Dieli-Conwright et al. (2021) examined MOTS-c responses to both aerobic and resistance exercise in Hispanic and non-Hispanic White breast cancer survivors, finding that exercise type differentially affected MOTS-c levels and that baseline MOTS-c was associated with metabolic health markers. The study was notable for examining MOTS-c in a clinical population rather than healthy volunteers, suggesting that exercise-induced MOTS-c production is preserved even in individuals with compromised health. It also introduced an ethnic dimension to MOTS-c research, as mitochondrial DNA (and therefore the MOTS-c coding sequence) varies by maternal lineage and population ancestry.[6]
Yoon et al. (2022) reviewed MOTS-c within the framework of exercise mitohormesis, the concept that exercise-induced mitochondrial stress produces beneficial adaptive responses. They positioned MOTS-c as a key mediator of this process: exercise stresses mitochondria, mitochondria release MOTS-c, MOTS-c activates AMPK and nuclear stress responses, and the cell adapts to become more metabolically efficient. This framing suggests MOTS-c is not merely an exercise byproduct but part of the signaling mechanism by which exercise produces its metabolic benefits.[7]
The exercise-mimetic framing requires honest qualification. MOTS-c replicates some of the metabolic effects of exercise (AMPK activation, glucose uptake, insulin sensitization) but not all of them. Exercise produces cardiovascular adaptations, neuromuscular coordination improvements, bone loading effects, psychological benefits, and neurotransmitter changes that no peptide can replicate. "Exercise mimetic" refers specifically to the metabolic signaling pathway, not to the full spectrum of exercise benefits.
The doubled running capacity in aged mice is the most frequently cited result, and it deserves context. The improvement occurred in mice that were sedentary at baseline. Whether MOTS-c would enhance exercise capacity in animals (or humans) that are already physically active is a different question. If MOTS-c's primary mechanism is restoring mitochondrial function that has declined with aging and inactivity, its benefits may be most apparent in the least active individuals, analogous to how exercise produces the largest metabolic benefits in previously sedentary people. For individuals already maintaining high physical activity levels, MOTS-c might provide diminishing returns. For the nuanced question of whether a peptide can substitute for physical activity, see MOTS-c and Physical Performance: Can a Peptide Replace a Workout?.
Aging, diabetes, and the decline of MOTS-c
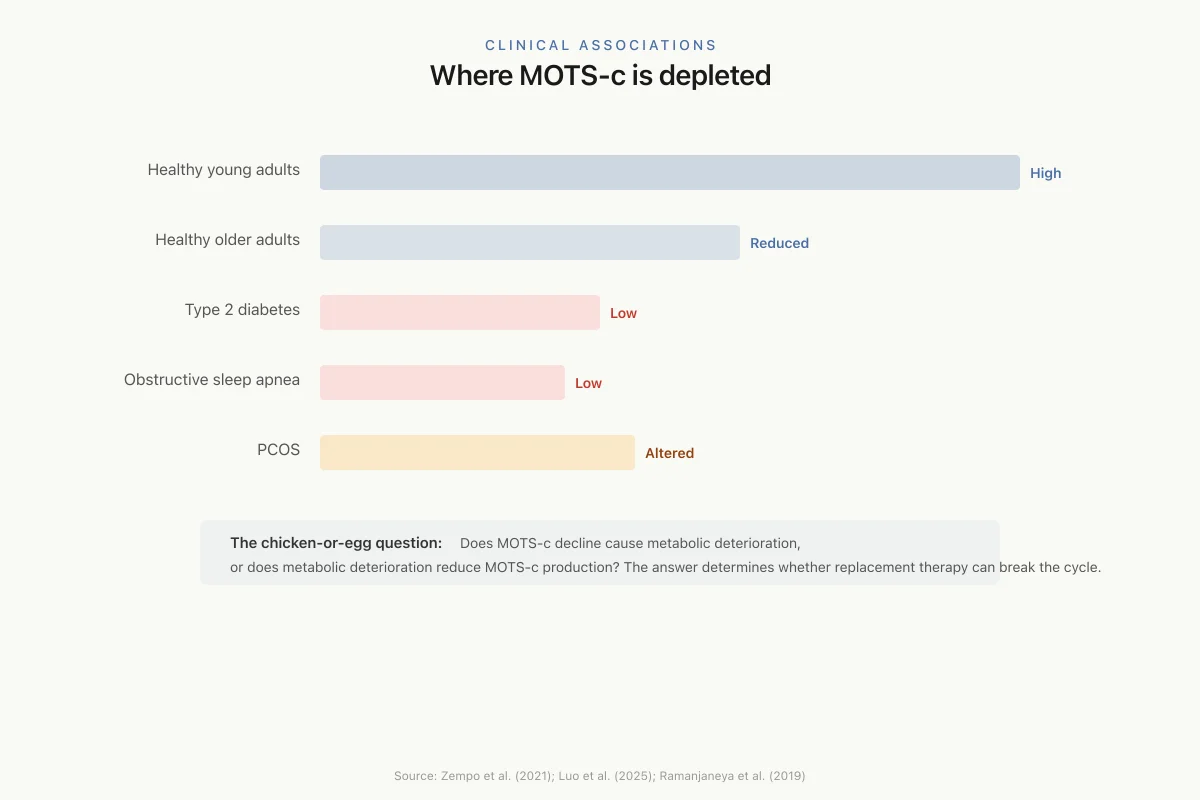
Circulating MOTS-c levels decline with age. This decline parallels the age-related reduction in mitochondrial function, insulin sensitivity, and physical capacity that characterizes metabolic aging. In humans, circulating MOTS-c levels are lower in patients with type 2 diabetes compared to healthy controls, suggesting a role in metabolic disease.
Zempo et al. (2021) identified a pro-diabetogenic mtDNA polymorphism (m.1382A>C) within the MOTS-c coding sequence that changes amino acid position 14 from lysine to glutamine. This variant was associated with increased risk of type 2 diabetes, providing genetic evidence that MOTS-c function is clinically relevant to metabolic health.[8]
Ramanjaneya et al. (2019) measured MOTS-c in patients with polycystic ovary syndrome (PCOS), finding that lipids and insulin regulate MOTS-c levels, and that the peptide was associated with metabolic parameters in both PCOS and healthy subjects.[9]
Luo et al. (2025) reported reduced serum MOTS-c levels in patients with obstructive sleep apnea, a condition associated with metabolic dysfunction and insulin resistance, further linking MOTS-c depletion to metabolic disease states.[10]
Clinical Associations
Where MOTS-c is depleted
The chicken-or-egg question: Does MOTS-c decline cause metabolic deterioration, or does metabolic deterioration reduce MOTS-c production? The answer determines whether replacing MOTS-c can break the cycle.
Source: Zempo et al. (2021); Luo et al. (2025); Ramanjaneya et al. (2019)
 View as image
View as imageThese findings create a narrative: MOTS-c is produced by healthy mitochondria, rises with exercise, declines with aging and metabolic disease, and when replaced, restores metabolic function in animal models. The analogy to other age-declining hormones (GH, testosterone, estrogen) is compelling but premature. The critical question is whether MOTS-c decline is a cause of metabolic aging or a consequence of it. If mitochondrial function declines first (due to accumulated mtDNA mutations, oxidative damage, or reduced mitochondrial biogenesis), then MOTS-c production declines as a downstream effect. Replacing MOTS-c without addressing the underlying mitochondrial dysfunction might provide temporary metabolic improvement without addressing root causes.
Alternatively, if MOTS-c itself is a key signal that maintains mitochondrial quality (through AMPK activation, which promotes mitochondrial biogenesis and autophagy), then its decline could initiate a vicious cycle: less MOTS-c means less AMPK activation means less mitochondrial biogenesis means less MOTS-c production. In this model, exogenous MOTS-c replacement could break the cycle and restore the positive feedback loop. The distinction matters for therapeutic strategy, and current evidence does not definitively favor either model. For the full context of age-related MOTS-c decline, see MOTS-c and Aging: Why Mitochondrial Peptides Decline with Age.
Toward clinical translation: the challenges
MOTS-c has not been tested as a therapeutic in human clinical trials. The primary barriers are pharmacokinetic: MOTS-c is a short peptide with low bioavailability, poor stability in plasma (rapid proteolytic degradation), a short half-life, and a tendency to persist at the injection site rather than distributing systemically.
CB4211, a more stable MOTS-c analog developed by CohBar Inc., completed Phase Ia/Ib clinical trials for non-alcoholic steatohepatitis (NASH) and obesity. The trial demonstrated a strong safety profile and preliminary efficacy in reducing liver fat content, providing the first evidence that MOTS-c-based therapeutics can produce measurable metabolic effects in humans. However, CohBar's development program faced financial and strategic challenges, and the broader clinical development timeline remains uncertain.
Wan et al. (2023) published a comprehensive review of MOTS-c's effects and mechanisms related to stress, metabolism, and disease, noting that while the preclinical evidence is robust, the translation to human therapeutics requires solving the delivery problem. Possible approaches include pegylation, lipidation, nanoparticle encapsulation, and the development of small-molecule MOTS-c mimetics that replicate the folate-methionine cycle inhibition without requiring peptide delivery.[5]
Zheng et al. (2023) reviewed the broader therapeutic potential, noting applications beyond metabolic disease in areas including cardiovascular protection, neuroprotection, and anti-inflammatory activity. The review emphasized that MOTS-c's mechanism (AMPK activation via folate cycle inhibition) is pharmacologically distinct from any existing drug class, representing a genuinely novel therapeutic approach if delivery challenges can be overcome.[11]
The delivery challenge is not trivial. MOTS-c is a 16-amino-acid peptide, small enough to be rapidly degraded by circulating proteases and cleared by the kidneys. In mouse studies, MOTS-c is typically administered by intraperitoneal injection at pharmacological doses (5-15 mg/kg/day) that far exceed endogenous circulating levels. Whether these mouse doses are translatable to human dosing, and whether chronic systemic administration can maintain the pulsatile, exercise-responsive pattern that characterizes endogenous MOTS-c signaling, are open questions.
The peptide's mitochondrial origin creates an additional complication for manufacturing. MOTS-c is encoded by mitochondrial DNA, which uses a slightly different genetic code than the nuclear genome (UGA encodes tryptophan in mitochondria rather than serving as a stop codon). This means MOTS-c must be produced by chemical synthesis rather than recombinant expression in bacterial or mammalian cell systems that use the standard genetic code. Chemical synthesis of a 16-amino-acid peptide is straightforward at laboratory scale but introduces cost and quality control challenges at pharmaceutical scale.
Lin et al. (2025) explored an alternative approach: creating MOTS-c-modified self-assembling peptide hydrogels that could provide sustained local delivery. This biomaterial strategy could bypass the systemic stability problem by delivering MOTS-c directly to target tissues, though it shifts the therapeutic application from systemic metabolic disease to local tissue applications like wound healing.
What the evidence does and does not show
The evidence for MOTS-c is strong in preclinical models and consistent in human observational data, but it lacks the clinical trial evidence that would support therapeutic claims. In mice, MOTS-c prevents obesity and insulin resistance (Lee 2015), doubles exercise capacity in aged animals (Reynolds 2021), and alters metabolic pathways in ways consistent with enhanced metabolic health (Kim 2019). In humans, MOTS-c is exercise-induced, declines with age, and is depleted in metabolic disease. The CB4211 analog showed preliminary efficacy in NASH.
The gaps are significant. No randomized controlled trial has tested MOTS-c itself in humans. The animal model data, while convincing, involves relatively short treatment durations and standardized mouse strains that may not reflect human metabolic diversity. The "exercise mimetic" label, while accurate in the narrow sense of AMPK activation and glucose uptake, overstates the breadth of effects when exercise produces dozens of beneficial adaptations that no single peptide can reproduce.
The mitochondrial origin of MOTS-c raises unique questions about genetic variation. Unlike nuclear-encoded peptides, MOTS-c is subject to maternal inheritance and mitochondrial DNA heteroplasmy (the presence of multiple mtDNA variants within a single cell). The Zempo et al. finding that a single nucleotide polymorphism in the MOTS-c coding region affects diabetes risk suggests that individual variation in MOTS-c sequence and expression could modify its therapeutic potential. A MOTS-c therapeutic developed from one mtDNA variant might not be equally effective in individuals carrying different variants. This is unlike nuclear-encoded peptide therapeutics, where the drug sequence is fixed and patient response depends on receptor variation. For MOTS-c, the patient's own mitochondria may be producing a slightly different version of the peptide, creating a pharmacogenomic variable that has no precedent in conventional drug development. The implications for personalized medicine are significant: a patient's mitochondrial haplotype could predict their response to MOTS-c-based therapy, but this type of pharmacogenomic screening has never been applied to a mitochondrial-derived peptide therapeutic.
The most recent research continues to expand MOTS-c's functional profile. Gudiksen et al. (2026) showed that MOTS-c improves intrinsic muscle mitochondrial bioenergetic health and efficiency in a PGC-1alpha-dependent manner, providing a direct mechanistic link between the peptide and mitochondrial quality.[12] This finding strengthens the positive feedback model: MOTS-c from mitochondria improves mitochondrial function, which would produce more MOTS-c, creating a self-reinforcing cycle of mitochondrial health.
Despite the limitations in current evidence, MOTS-c represents a conceptually important advance. It demonstrates that mitochondria encode bioactive peptides that regulate systemic metabolism, that these peptides are exercise-responsive, and that their decline with aging contributes to metabolic deterioration. Whether this biology can be harnessed therapeutically depends on solving the delivery problem and conducting the human trials that the preclinical data justifies. The fact that exercise naturally induces MOTS-c production provides a non-pharmacological strategy that is available today: physical activity may be the most effective way to maintain endogenous MOTS-c levels and the metabolic benefits this peptide confers.
The Bottom Line
MOTS-c is a 16-amino-acid mitochondrial-derived peptide that activates AMPK, enhances insulin sensitivity, and mimics key metabolic effects of exercise. Discovered in 2015, it has strong preclinical evidence for preventing obesity and insulin resistance in mice and doubling exercise capacity in aged animals. In humans, MOTS-c levels rise 12-fold with exercise and decline with aging and diabetes. No human therapeutic trial has been completed, though a stabilized analog (CB4211) showed preliminary efficacy in NASH. The delivery challenge (low stability, short half-life) remains the primary barrier to clinical development.
Sources & References
- 1RPEP-02703·Lee, Changhan et al. (2015). “The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance..” Cell metabolism.Study breakdown →PubMed →↩
- 2RPEP-05720·Reynolds, Joseph C et al. (2021). “MOTS-c: The Mitochondrial Peptide That Mimics Exercise and Fights Aging — Even When Started Late in Life.” Nature communications.Study breakdown →PubMed →↩
- 3RPEP-03003·Lee, Changhan et al. (2016). “MOTS-c: A novel mitochondrial-derived peptide regulating muscle and fat metabolism..” Free radical biology & medicine.Study breakdown →PubMed →↩
- 4RPEP-04280·Kim, Su-Jeong et al. (2019). “The mitochondrial-derived peptide MOTS-c is a regulator of plasma metabolites and enhances insulin sensitivity..” Physiological reports.Study breakdown →PubMed →↩
- 5RPEP-07506·Wan, Wei et al. (2023). “MOTS-c: The Mitochondrial Peptide That Links Exercise, Metabolism, and Aging.” Journal of translational medicine.Study breakdown →PubMed →↩
- 6RPEP-05345·Dieli-Conwright, Christina M et al. (2021). “Exercise Boosts the Mitochondrial Peptide MOTS-c in Breast Cancer Survivors.” Scientific reports.Study breakdown →PubMed →↩
- 7RPEP-06635·Yoon, Tae Kwan et al. (2022). “Exercise, Mitohormesis, and Mitochondrial ORF of the 12S rRNA Type-C (MOTS-c)..” Diabetes & metabolism journal.Study breakdown →PubMed →↩
- 8RPEP-05924·Zempo, Hirofumi et al. (2021). “A pro-diabetogenic mtDNA polymorphism in the mitochondrial-derived peptide, MOTS-c..” Aging.Study breakdown →PubMed →↩
- 9RPEP-04440·Ramanjaneya, Manjunath et al. (2019). “Lipids and insulin regulate mitochondrial-derived peptide (MOTS-c) in PCOS and healthy subjects..” Clinical endocrinology.Study breakdown →PubMed →↩
- 10RPEP-12342·Luo, Zhuoding et al. (2025). “People with Worse Sleep Apnea Have Lower Levels of the Mitochondrial Peptide MOTS-c.” Sleep and biological rhythms.Study breakdown →PubMed →↩
- 11RPEP-07641·Zheng, Yuejun et al. (2023). “MOTS-c: A Tiny 16-Amino-Acid Peptide from Mitochondria That Could Treat Diabetes, Obesity, and Aging.” Frontiers in endocrinology.Study breakdown →PubMed →↩
- 12RPEP-15235·Gudiksen, Anders et al. (2026). “MOTS-c Peptide Improves Muscle Energy Production Efficiency.” Free radical biology & medicine.Study breakdown →PubMed →↩