Compounded Peptide Safety: The Regulatory Gap
Peptide Safety Monitoring
900+ Adverse Event Reports
By December 2024, the FDA's FAERS database contained over 900 adverse event reports linked to compounded semaglutide and tirzepatide, including 17 deaths. A 2026 pharmacovigilance study found compounded GLP-1 products had 2.35x higher hospitalization odds.
McCall et al., Expert Opin Drug Saf, 2026
McCall et al., Expert Opin Drug Saf, 2026
If you only read one thing
FDA-approved peptide drugs like Ozempic go through clinical trials and are tracked for side effects after they hit the market. Compounded versions of those same drugs skip most of that. A 2026 study looking at government safety reports found compounded GLP-1 products had over twice the odds of sending someone to the hospital, nearly 50 times the odds of preparation errors, and 19 times the odds of contamination. For peptides like BPC-157 and TB-500, it's worse: no approved product exists to compare against, and no one is systematically tracking safety at all.
The peptide market operates across two parallel systems with fundamentally different safety oversight. FDA-approved peptide drugs (semaglutide as Ozempic/Wegovy, tirzepatide as Mounjaro/Zepbound) pass through clinical trials with thousands of participants, post-marketing surveillance via the FAERS database, and continuous manufacturing inspections. Compounded peptides, whether from 503A pharmacies, 503B outsourcing facilities, or gray-market "research chemical" vendors, face a patchwork of oversight that ranges from inconsistent to nonexistent. A 2026 pharmacovigilance analysis of the FDA's FAERS database found that compounded GLP-1 receptor agonists had 2.35 times higher odds of hospitalization compared to their FDA-approved counterparts.[1] This article maps the regulatory landscape, the safety signals that have emerged, and the structural gaps that allow compounded peptides to reach consumers with minimal monitoring. For context on how these regulatory dynamics affect specific peptides, see BPC-157 and the FDA: The Category 2 Classification Explained.
Key Takeaways
- Compounded GLP-1 receptor agonists showed 2.35x higher odds of hospitalization vs. FDA-approved versions in a FAERS analysis of 81,078 reports (McCall et al., Expert Opin Drug Saf, 2026)
- Preparation errors were 48.9x more likely with compounded products, and contamination reports were 19x higher (McCall et al., 2026)
- The FDA classified BPC-157, TB-500, CJC-1295, Ipamorelin, and 10+ other peptides as Category 2 in September 2023, effectively barring them from legal compounding
- By December 2024, FAERS contained 900+ adverse event reports from compounded semaglutide and tirzepatide, including 17 deaths
- No dedicated post-marketing surveillance system exists for compounded peptides; the FAERS system is voluntary and captures only a fraction of actual adverse events
- A 2025 narrative review found only three human pilot studies in the entire BPC-157 literature, with no clinical safety data (McGuire et al., 2025)
How the Two-Track System Works
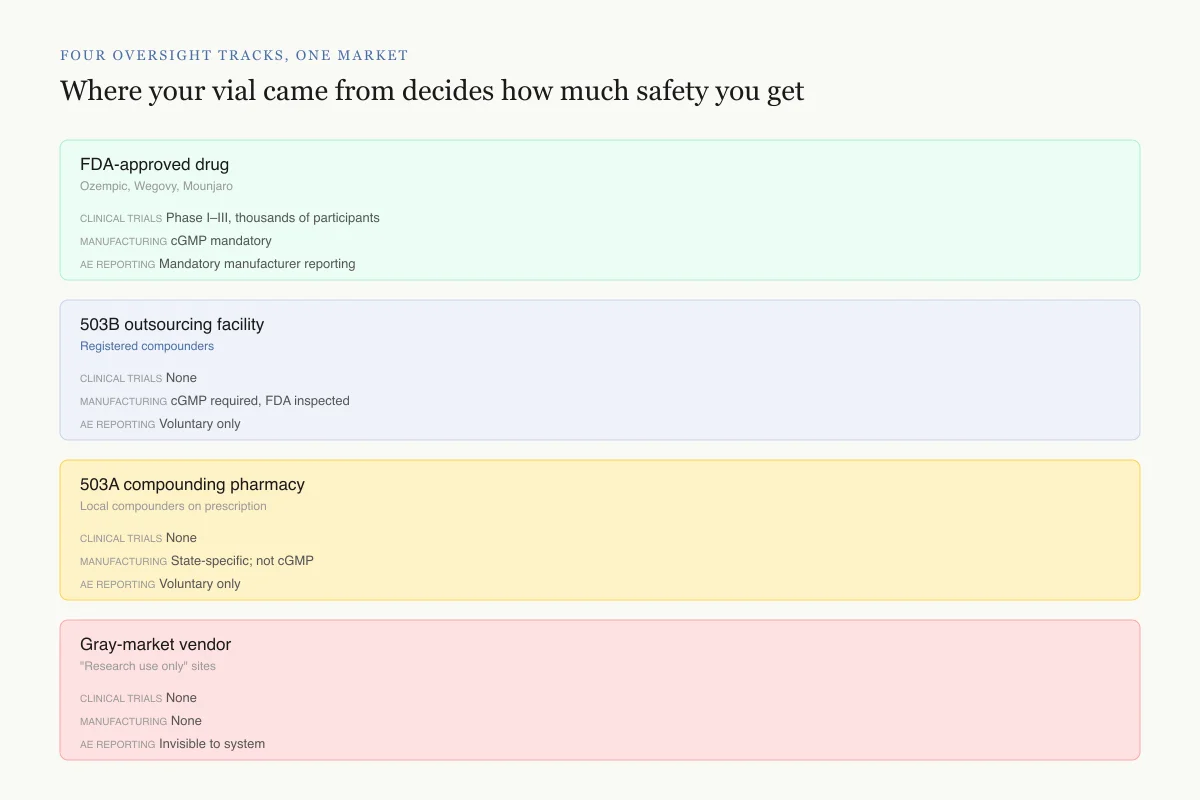
Four oversight tracks, one market
Where your vial came from decides how much safety you get
FDA-approved drug
Ozempic, Wegovy, Mounjaro
Clinical trials
Phase I–III, thousands of participants
Manufacturing standards
cGMP mandatory
Adverse event reporting
Mandatory manufacturer reporting
503B outsourcing facility
Registered compounders
Clinical trials
None
Manufacturing standards
cGMP required, FDA inspected
Adverse event reporting
Voluntary only
503A compounding pharmacy
Local compounders on prescription
Clinical trials
None
Manufacturing standards
State-specific; not cGMP
Adverse event reporting
Voluntary only
Gray-market vendor
"Research use only" sites
Clinical trials
None
Manufacturing standards
None
Adverse event reporting
Invisible to system
All four tracks sell products marketed under the same peptide name. Three of them generate no pre-market safety data, and all three compounded tracks have no mandatory adverse event reporting.
Source: FDA compounding framework (sections 503A, 503B of FDCA); FAERS reporting rules
 View as image
View as imageTrack 1: FDA-Approved Peptide Drugs
FDA-approved peptide drugs follow the standard pharmaceutical pathway. Before reaching patients, they pass through Phase I (safety in healthy volunteers), Phase II (efficacy and dosing), and Phase III (large-scale efficacy and safety) clinical trials. After approval, the manufacturer must report adverse events, and the FDA monitors the FAERS database for safety signals.
This system has known limitations. FAERS is a voluntary reporting system, meaning most adverse events go unreported. Estimates suggest FAERS captures between 1% and 10% of actual adverse events. For rare side effects that take months or years to manifest, the system is slow. But it exists, and it produces actionable data. The GLP-1 receptor agonist class has generated tens of thousands of FAERS reports, enabling pharmacovigilance studies that have identified signals for pancreatitis, gallbladder disease, gastroparesis, and potential ocular effects. For how these safety signals are identified and evaluated, see Peptide Post-Marketing Surveillance: How Safety Signals Are Detected.
Track 2: Compounded Peptide Products
Compounded medications occupy a legal space carved out by sections 503A and 503B of the Federal Food, Drug, and Cosmetic Act. The original intent was to allow pharmacies to customize medications for individual patients (different doses, alternative delivery forms, allergen-free formulations) when a commercially available product does not meet a specific patient need.
503A pharmacies are traditional compounding pharmacies that prepare drugs based on individual prescriptions. They must be state-licensed but face limited federal oversight. They cannot compound from bulk drug substances that the FDA has identified as presenting "significant safety risks" (Category 2 substances).
503B outsourcing facilities register voluntarily with the FDA, follow current Good Manufacturing Practices (cGMP), and can compound without individual prescriptions. They face more oversight than 503A pharmacies but less than traditional pharmaceutical manufacturers.
Gray-market vendors sell peptides labeled "for research use only" or "not for human consumption." These products have no regulatory oversight, no manufacturing standards, no purity verification, and no adverse event reporting. They are technically outside the compounding framework entirely.
For the legal fiction underlying this market, see "For Research Use Only": The Legal Fiction of Gray-Market Peptides.
The Category 2 Classification
In September 2023, the FDA updated its list of bulk drug substances that may present significant safety risks when used in compounding. The Category 2 designation means the substance has been evaluated and the FDA has identified safety concerns that effectively bar it from compounding under both 503A and 503B frameworks.
Peptides classified as Category 2 include:
- BPC-157 (Body Protection Compound 157)
- TB-500 (Thymosin beta-4 fragment)
- AOD-9604 (growth hormone fragment)
- CJC-1295 (growth hormone releasing hormone analog)
- Ipamorelin (growth hormone secretagogue)
- Melanotan II (melanocortin receptor agonist)
- Selank and Semax (synthetic peptide nootropics)
- KPV (alpha-MSH fragment)
The FDA cited four primary safety concerns across these substances: limited or inadequate safety data, concerns about characterization and impurities, lack of evidence of historical use in compounding, and potential immunogenicity risks. For BPC-157 specifically, a 2025 systematic review found 544 published articles but only 1 clinical study among 36 that met inclusion criteria, and no clinical safety data.[2] A separate 2025 narrative review identified only three human pilot studies in the entire BPC-157 literature.[3]
Category 1 substances (permitted for compounding) include a much shorter peptide list: GHK-Cu (with limitations) and vasoactive intestinal peptide. The vast majority of popular "wellness peptides" are either Category 2 (restricted) or have not been nominated for evaluation at all.
The Compounded GLP-1 Safety Data
FAERS Pharmacovigilance · McCall 2026
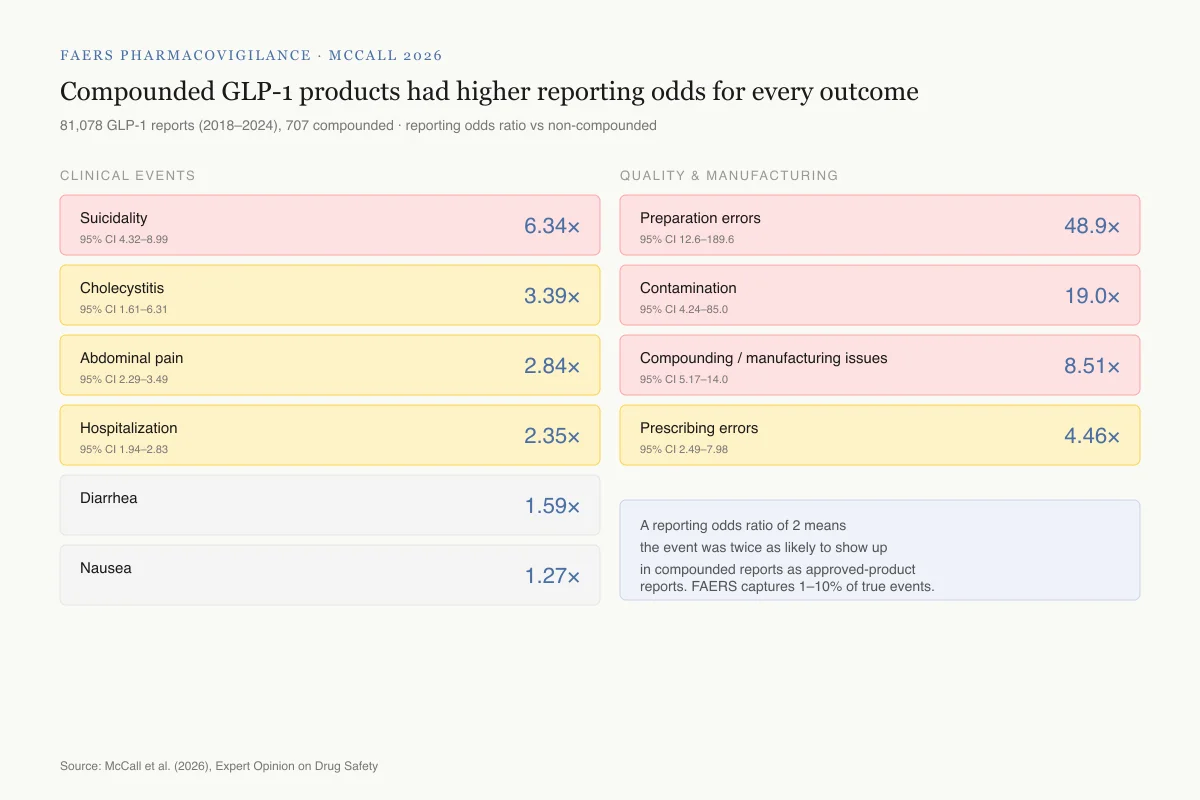
Compounded GLP-1 products had higher reporting odds for every outcome measured
81,078 GLP-1 receptor agonist reports (2018–2024), 707 involving compounded products · reporting odds ratio vs non-compounded
Clinical events
Suicidality
95% CI 4.32–8.99
6.34×
Cholecystitis
95% CI 1.61–6.31
3.39×
Abdominal pain
95% CI 2.29–3.49
2.84×
Hospitalization
95% CI 1.94–2.83
2.35×
Diarrhea
95% CI 1.25–1.99
1.59×
Nausea
95% CI 1.05–1.52
1.27×
Quality and manufacturing
Preparation errors
95% CI 12.6–189.6
48.9×
Contamination
95% CI 4.24–85.0
19.0×
Compounding / manufacturing issues
95% CI 5.17–14.0
8.51×
Prescribing errors
95% CI 2.49–7.98
4.46×
Reading the numbers: A reporting odds ratio of 2 means the adverse event was twice as likely to appear in compounded reports as in approved-product reports. FAERS captures 1–10% of true events, so these are comparisons of the tip of the iceberg.
Source: McCall et al. (2026), Expert Opinion on Drug Safety
 View as image
View as imageThe most comprehensive safety comparison between compounded and FDA-approved peptides comes from McCall et al. (2026), published in Expert Opinion on Drug Safety.[1] They analyzed 81,078 GLP-1 receptor agonist reports in the FAERS database from 2018 to 2024, of which 707 involved compounded products.
Adverse Event Differences
Compounded GLP-1 products showed significantly higher reporting odds ratios (RORs) compared to non-compounded versions:
| Adverse Event | Compounded ROR (95% CI) |
|---|---|
| Suicidality | 6.34 (4.32-8.99) |
| Cholecystitis | 3.39 (1.61-6.31) |
| Abdominal pain | 2.84 (2.29-3.49) |
| Hospitalization | 2.35 (1.94-2.83) |
| Diarrhea | 1.59 (1.25-1.99) |
| Nausea | 1.27 (1.05-1.52) |
The suicidality signal (ROR 6.34) is striking, though the absolute numbers remain small and the FAERS data cannot establish causation. The hospitalization odds (2.35x) are more immediately concerning from a public health standpoint.
Manufacturing and Quality Signals
The manufacturing-related findings were even more dramatic:
| Quality Issue | Compounded ROR (95% CI) |
|---|---|
| Preparation errors | 48.92 (12.63-189.6) |
| Contamination | 19.00 (4.24-85.03) |
| Compounding/manufacturing issues | 8.51 (5.17-14.0) |
| Prescribing errors | 4.46 (2.49-7.98) |
Preparation errors were nearly 49 times more likely with compounded products. The wide confidence interval (12.63-189.6) reflects the relatively small number of reports, but the direction is unambiguous. Contamination was 19 times more likely. These are not pharmacological differences between the peptide molecules; they are manufacturing and quality control failures.
One counterintuitive finding: compounded products showed lower odds of administration errors (0.29) and dosing errors (0.24). This likely reflects the different patient populations. FDA-approved GLP-1 drugs are prescribed to millions of patients, many using self-injection pens for the first time. Compounded products reach a smaller, often more experienced population through clinics and telemedicine providers.
What the Numbers Miss
The McCall study has important limitations that likely understate the actual safety differential. FAERS is a voluntary reporting system, and the denominator (total users) is unknown for both compounded and non-compounded products. If compounded GLP-1 users are less likely to report adverse events (due to less established provider relationships, out-of-pocket payment without insurance tracking, or reluctance to report problems with products obtained through non-traditional channels), then the true adverse event rate for compounded products could be higher than the reported odds ratios suggest.
Additionally, the study period (2018-2024) coincides with the semaglutide shortage, during which compounded GLP-1 products were prescribed at unprecedented volume, often to patients who could not access the approved product. Whether the safety signals reflect inherent product quality problems, the rushed scaling of compounding operations during the shortage, or both, cannot be separated in the FAERS data.
The 17 deaths reported in association with compounded semaglutide and tirzepatide by December 2024 represent cases where compounded products were identified in FAERS. Given the limitations of the reporting system, the actual number of fatalities associated with compounded GLP-1 products during this period is unknown. Some deaths may have been attributed to the active ingredient rather than the compounded formulation, and some may never have been reported to FAERS at all.
For a broader perspective on how BPC-157's safety profile compares to approved peptide therapeutics, see BPC-157: The Body Protection Compound and What the Research Shows.
Where the Monitoring Fails
No Dedicated System for Compounded Products
FAERS was designed to monitor FDA-approved drugs. Compounded preparations are technically reportable, but in practice, they are underreported for several reasons:
Identification problems: Compounded products do not have National Drug Codes (NDCs), making them difficult to track in electronic health records and adverse event reporting systems. A patient who experiences an adverse event from compounded semaglutide may have that event attributed to "semaglutide" generically, without distinguishing it from Ozempic or Wegovy.
Reporting incentives: Pharmaceutical manufacturers are legally required to report adverse events for their products. Compounding pharmacies have no equivalent reporting obligation. Clinics that prescribe compounded peptides face no systematic requirement to track outcomes.
Patient barriers: Many compounded peptide users obtain products through telehealth clinics or direct purchase from gray-market vendors. These patients may not have an established relationship with a healthcare provider who would recognize and report an adverse event. Some may not associate a delayed side effect with their peptide use.
The Gray Market Is Invisible
The FAERS data captures only the regulated compounding market (503A and 503B pharmacies). Gray-market peptides, sold as "research chemicals" with no quality controls, generate no safety data at all. State attorneys general have documented consumers mixing raw peptide powders at home, leading to overdoses and contamination exposures. These events are invisible to the pharmacovigilance system.
The scale of gray-market peptide use is unknown. Google search volume for peptides like BPC-157, TB-500, and various growth hormone secretagogues runs into hundreds of thousands of monthly searches. Reddit forums, TikTok, and YouTube contain detailed user protocols with dosing, reconstitution, and injection instructions. None of this generates reportable safety data. For the full picture of BPC-157's research-to-consumer pipeline, see The Real BPC-157 Story: 544 Papers, 30 Human Subjects, 50 Million Views.
Immunogenicity Is Unmeasured
Peptides can trigger immune responses, particularly when injected. The immune system may recognize a synthetic peptide as foreign, generating anti-drug antibodies (ADAs) that neutralize the peptide, reduce its efficacy, or cause allergic reactions. Immunogenicity risk increases with impurities, aggregation (peptide molecules clumping together), and degradation products, all of which are more likely in compounded formulations with less rigorous manufacturing controls.
For FDA-approved peptide drugs, immunogenicity is extensively characterized during clinical trials, with specific assays measuring ADA formation. For compounded peptides, immunogenicity is neither measured nor monitored. A 2025 review noted that even trace levels of residual solvents or synthesis by-products in compounded peptides could trigger adverse immune responses, particularly with repeated injections over time. This risk is theoretical for most compounded peptides because the studies to measure it have simply never been conducted.
The Specific Problem with Non-GLP-1 Peptides
The McCall 2026 study focused on compounded GLP-1 receptor agonists, which at least have FDA-approved reference products (Ozempic, Wegovy, Mounjaro) enabling comparison. For peptides like BPC-157, TB-500, and growth hormone secretagogues, the safety monitoring gap is fundamentally wider because no approved reference product exists.
When a patient uses compounded semaglutide and experiences an adverse event, the pharmacovigilance system can at least contextualize it against the known safety profile of approved semaglutide. When a patient uses BPC-157 and experiences a problem, there is no approved safety profile to compare against. The 2025 Vasireddi systematic review found that BPC-157 preclinical safety studies showed no adverse effects across several organ systems, but "no clinical safety data were found."[2] The McGuire 2025 review reached the same conclusion: "adverse effects are possible due to unregulated manufacturing, contamination, or unknown clinical safety."[3]
This means that for Category 2 peptides, three independent layers of safety assurance are simultaneously absent:
- No pre-market clinical safety data from controlled human trials
- No standardized manufacturing to ensure purity and consistency
- No post-market surveillance to detect problems that emerge with widespread use
Each missing layer compounds the risk of the others. Without clinical trials, the safe dose range is unknown. Without manufacturing standards, the actual dose delivered is uncertain. Without surveillance, emerging problems remain invisible until they become severe enough to generate spontaneous reports or media attention.
The Sports Anti-Doping Signal
The World Anti-Doping Agency (WADA) banned BPC-157, TB-500, and several other non-approved peptides from competitive sports. The NFL, UFC, and NCAA followed. These bans were motivated by performance-enhancement concerns rather than safety data, but they produced an unintended pharmacovigilance signal: athletes testing positive for these substances demonstrated that compounded peptides are being used at scale in populations that undergo regular medical monitoring. Yet even in this monitored population, no systematic safety tracking occurs. Anti-doping tests detect the substance; they do not record health outcomes.
The Regulatory Timeline
How we got here
The compounded peptide regulatory timeline
Each event shapes what oversight compounded peptides do or don't receive today.
2012
The NECC outbreak
Contaminated compounded steroid injections kill 76 people, sicken 750+. Congress responds with the Drug Quality and Security Act.
2013
503B framework created
DQSA establishes voluntary 503B outsourcing facilities with cGMP requirements and FDA inspection.
2013–2022
Wellness peptide market grows
BPC-157, TB-500, CJC-1295, Ipamorelin, and similar peptides sold widely through clinics and gray-market vendors with little intervention.
Sept 2023
Category 2 expansion
FDA classifies BPC-157, TB-500, AOD-9604, CJC-1295, Ipamorelin, Melanotan II, Selank, Semax, KPV as Category 2 — effectively banned from legal compounding.
2024
GLP-1 shortage fuels compounding surge
Semaglutide and tirzepatide shortages allow mass compounding. By December, FAERS logs 900+ adverse events and 17 deaths linked to compounded GLP-1 products.
Feb 2025
Shortage declared resolved
FDA ends compounding allowance. Enforcement actions begin. Legal challenges from 503B facilities follow.
2026
McCall pharmacovigilance study
Analysis of 81,078 FAERS reports shows compounded GLP-1s carry 2.35× higher hospitalization odds and 48.9× higher preparation-error odds.
Source: FDA compounding policy history; McCall et al. (2026); DQSA legislative record
 View as image
View as imageThe current regulatory landscape emerged through a series of events:
2012: The Drug Quality and Security Act created the 503B outsourcing facility framework after a compounded steroid injection contamination outbreak killed 76 people and sickened over 750.
2013-2022: The "wellness peptide" market grew largely without FDA intervention. Compounding pharmacies and clinics offered BPC-157, TB-500, growth hormone secretagogues, and other peptides to a growing consumer base.
September 2023: FDA added multiple peptides to the Category 2 bulk drug substances list, effectively restricting them from legal compounding.
2024-2025: The national GLP-1 drug shortage created massive demand for compounded semaglutide. The FDA allowed compounding during the shortage but signaled enforcement would follow when supply stabilized.
February 2025: FDA clarified that the semaglutide shortage had resolved, triggering enforcement actions against compounders. State attorneys general also began actions against compounded GLP-1 sellers.
2026: Enforcement intensified, with the FDA and state regulators targeting both compounding pharmacies and gray-market sellers. For information on how the BPC-157 classification specifically works, see BPC-157 and the FDA: The Category 2 Classification Explained.
What the Gap Means in Practice
The regulatory gap between approved and compounded peptides creates several concrete problems:
No dose-response data: For most compounded peptides, the effective dose, toxic dose, and therapeutic window have not been established in humans. Users extrapolate from animal studies or anecdotal reports. The McGuire 2025 review of BPC-157 concluded that "until well-designed clinical trials are conducted, BPC-157 should be considered investigational."[3]
No drug interaction data: Many peptide users combine multiple peptides simultaneously ("stacking") or use peptides alongside prescription medications. No interaction studies exist for these combinations.
No long-term safety data: Even for peptides with extensive animal research, long-term human safety data is absent. Animal studies of BPC-157 typically run 14-42 days. Human users often take compounded peptides for months or years. For the full scope of what remains unknown about long-term peptide use, see Long-Term Peptide Safety Data Gaps: What We Still Don't Know.
Purity is variable: Without standardized manufacturing and testing, compounded peptide purity varies by source. Third-party testing services exist but are not required, and their quality varies. A vial labeled "BPC-157 5mg" from one source may contain a different quantity, different purity, or different peptide than the same label from another source.
Batch-to-batch inconsistency: Even within a single compounding pharmacy, different batches of the same peptide may have different potency, purity, and stability profiles. FDA-approved drugs undergo batch release testing; compounded preparations may or may not.
The Path Forward: What Would Close the Gap
Closing the regulatory gap between approved and compounded peptides would require action at multiple levels, none of which is currently underway in a comprehensive form.
Mandatory adverse event reporting for compounding pharmacies: Currently, compounders have no federal reporting obligation equivalent to what pharmaceutical manufacturers face. Requiring 503A and 503B facilities to report adverse events to FAERS would at minimum make safety signals visible.
National Drug Codes for compounded peptides: Assigning trackable identifiers to compounded preparations would allow health systems and pharmacovigilance databases to distinguish compounded from approved products, eliminating the identification problem that currently obscures safety data.
Clinical trials for commonly used peptides: The FDA's Category 2 classification is based on insufficient safety data. The straightforward solution is generating that data. For peptides like BPC-157 with over 500 published preclinical studies, the research community has the mechanistic foundation to design targeted human trials. The barrier is not scientific feasibility but economic incentive: peptides without patent protection offer limited commercial return on clinical trial investment.
Standardized third-party testing: An independent testing and certification program for compounded peptides, similar to USP verification for supplements, could provide quality assurance without requiring FDA approval. This would not address efficacy questions but could reduce contamination and potency variation.
Consumer education: Many peptide users do not understand the difference between FDA-approved drugs, legally compounded preparations from 503A or 503B pharmacies, and gray-market research chemicals sold online without any quality oversight or regulatory accountability. Clearer labeling requirements and accessible consumer-facing information about the regulatory status, manufacturing origin, and testing history of their specific products would enable more informed decision-making about the actual risk profile of what they are injecting.
None of these measures would eliminate risk. But the current system, where compounded peptides reach consumers through multiple channels with no standardized quality control and no systematic safety monitoring, represents the widest gap between pharmaceutical regulation and actual market practice in modern medicine.
Safety
CriticalGray-market peptides carry risks that don't show up in any safety database
Concern
Products labeled 'research use only' have no manufacturing standards, no purity verification, and no adverse event tracking. Contamination, wrong dose, wrong peptide, and unknown impurities are all documented problems. Immunogenicity — the risk of triggering an immune reaction, especially with repeated injection of impure peptides — is neither measured nor monitored.
What the research says
If you're using a peptide for a wellness goal, the safest compounded source is a 503B outsourcing facility prescribed by a clinician who can follow up on side effects. Gray-market 'research chemical' vendors sit at the opposite end of the spectrum. For Category 2 peptides like BPC-157, even the legal compounded route is now closed in the US.
Particularly relevant for: Anyone considering peptides outside FDA-approved products
What to do
Verify the source's regulatory track. Ask for third-party purity testing on the exact lot you're receiving. Report any unexplained symptoms to FDA MedWatch so the signal reaches FAERS. Never self-inject a 'research' product.
FDA compounding framework; Vasireddi 2025; McCall 2026 FAERS analysis
For an overview of what remains unknown about the long-term effects of peptide use, see Long-Term Peptide Safety Data Gaps: What We Still Don't Know.
The Bottom Line
Compounded peptides operate in a regulatory space with minimal safety monitoring compared to FDA-approved drugs. A 2026 FAERS analysis found compounded GLP-1 products had 2.35x higher hospitalization odds, 49x higher preparation error rates, and 19x higher contamination reports. The FDA's Category 2 classification restricts many popular peptides from legal compounding, but gray-market sales continue without any safety monitoring. No post-marketing surveillance system exists specifically for compounded peptide products, and the gap between animal research and human safety data remains the central unresolved challenge in peptide regulation.
Sources & References
- 1RPEP-15689·McCall, Kenneth L et al. (2026). “Compounded GLP-1 Drugs Show Dramatically Higher Rates of Side Effects and Quality Problems Than FDA-Approved Versions.” Expert opinion on drug safety.Study breakdown →PubMed →↩
- 2RPEP-13892·Vasireddi, Nikhil et al. (2025). “BPC-157 for Sports Injuries: What a Systematic Review of the Evidence Actually Shows.” HSS journal : the musculoskeletal journal of Hospital for Special Surgery.Study breakdown →PubMed →↩
- 3RPEP-12503·McGuire, Flynn P et al. (2025). “BPC-157 for Muscle and Tendon Healing: What Science Actually Shows vs. What Athletes Believe.” Current reviews in musculoskeletal medicine.Study breakdown →PubMed →↩