How Semax Upregulates BDNF: The Mechanism
Semax
1.4x BDNF
A single intranasal dose of Semax produces a 1.4-fold increase in BDNF protein and a 3-fold increase in BDNF mRNA in the rat hippocampus, with peak TrkB phosphorylation rising 1.6-fold.
Dolotov et al., Brain Research, 2006
Dolotov et al., Brain Research, 2006
If you only read one thing
Semax is a nasal spray peptide from Russia that boosts BDNF — the main brain protein responsible for forming new neural connections and memories. One dose raised BDNF protein by 40% and BDNF gene activity by 3x in the hippocampus (the brain's memory center). It also activates the receptor that BDNF binds to, so it amplifies the signal at both ends. The catch: this is all from rat studies. The data is strong and consistent at the molecular level, but it hasn't been validated in controlled human trials outside Russia.
Semax (Met-Glu-His-Phe-Pro-Gly-Pro) is a synthetic heptapeptide based on the ACTH(4-10) fragment. Developed at the Institute of Molecular Genetics in Moscow during the 1980s, it was designed to retain the neurotrophic properties of ACTH while eliminating its hormonal (steroidogenic) activity. The most consistently documented molecular effect of Semax is upregulation of brain-derived neurotrophic factor (BDNF) and its receptor TrkB across multiple brain regions.[1] This BDNF-TrkB pathway is the primary molecular substrate of synaptic plasticity, the process by which neural connections strengthen or weaken in response to experience.
For a deeper look at BDNF itself, its discovery, and its role across neurological conditions, see BDNF: The Brain Peptide That Builds New Neural Connections. For the broader family of Russian-developed peptide bioregulators, see Cortexin: The Brain Peptide Bioregulator from Russian Medicine.
Key Takeaways
- Semax is a nasal spray that bumps up BDNF — the brain's main "build new connections" protein.
- A single dose in rats raised BDNF protein 40% and tripled its gene activity in the memory center.
- It also boosts the receptor that catches BDNF — so the signal gets amplified at both ends.
- It was built from a piece of the stress hormone ACTH, stripped of the part that raises cortisol.
- Brain gene changes start within 20 minutes of a single dose of Semax.
- It specifically turns up the survival signal without touching the cell-death pathway right next to it.
- Nearly all Semax evidence comes from rats and Russian labs — no Western controlled human trials exist.
What Semax Is and How It Relates to ACTH
Semax consists of the ACTH(4-7) core sequence (Met-Glu-His-Phe) with an added Pro-Gly-Pro tripeptide at the C-terminus. The PGP tail serves two purposes: it increases resistance to enzymatic degradation, extending the peptide's biological half-life, and it may contribute its own biological activity. The tripeptide Pro-Gly-Pro is itself a collagen fragment with documented immunomodulatory properties.
The critical design principle was separating ACTH's neurotrophic effects from its endocrine effects. Full-length ACTH (39 amino acids) stimulates the adrenal cortex to produce cortisol. The 4-10 fragment retains cognitive and neurotrophic activity without stimulating steroidogenesis. Early studies in the 1990s at the Institute of Molecular Genetics confirmed this separation: Semax enhanced learning and memory in animal models without altering corticosteroid levels.[7]
Semax is administered intranasally. This route bypasses the blood-brain barrier by exploiting olfactory and trigeminal nerve pathways, allowing direct access to the central nervous system. For the science behind this delivery method, see Semax Nasal Delivery: How It Reaches the Brain.
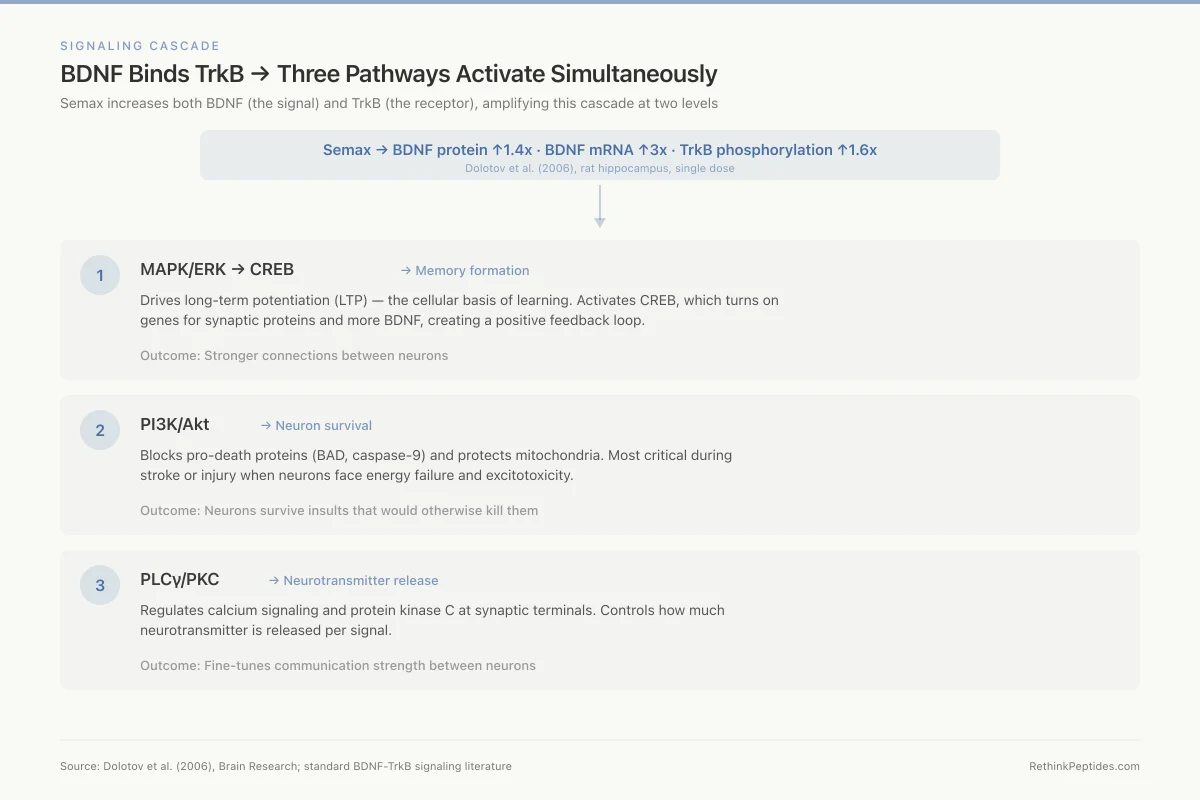
The BDNF-TrkB Signaling Cascade
BDNF is the most abundant neurotrophin in the adult brain. It is synthesized as a precursor protein (proBDNF), which is cleaved to produce the mature 12.3 kDa form that binds TrkB. The precursor and mature forms have opposing effects: proBDNF binds the p75NTR receptor and can trigger apoptosis, while mature BDNF binds TrkB and promotes survival and plasticity. The balance between these two forms is itself a regulatory mechanism, and any intervention that increases total BDNF must be evaluated for which form predominates.
When mature BDNF binds its high-affinity receptor TrkB (tropomyosin receptor kinase B), TrkB dimerizes and autophosphorylates specific tyrosine residues in its intracellular domain. This phosphorylation creates docking sites for adapter proteins that initiate three major intracellular signaling cascades:
- MAPK/ERK pathway controls gene expression for synaptic proteins and drives long-term potentiation (LTP), the cellular mechanism of memory formation. ERK phosphorylation activates the transcription factor CREB (cAMP response element-binding protein), which in turn drives expression of genes encoding synaptic structural proteins, ion channels, and BDNF itself, creating a positive feedback loop.
- PI3K/Akt pathway promotes neuronal survival by phosphorylating and inhibiting pro-apoptotic proteins (BAD, caspase-9) and maintaining mitochondrial membrane integrity. This pathway is particularly relevant during ischemic stress, when neurons face energy failure and excitotoxicity.
- PLC-gamma/PKC pathway hydrolyzes membrane phospholipids to generate IP3 and DAG, which regulate intracellular calcium release and protein kinase C activation. These second messengers modulate neurotransmitter release probability at the presynaptic terminal and contribute to short-term synaptic plasticity.
These cascades converge on outcomes that define neuroplasticity: new synaptic connections form, existing connections strengthen, neurons survive insults that would otherwise kill them, and adult neurogenesis continues in the hippocampal dentate gyrus. The convergence point is gene expression. All three pathways ultimately alter which proteins a neuron produces, shifting its molecular identity toward a more plastic, more resilient state.
BDNF-TrkB signaling is not a simple on/off switch. The magnitude, duration, and spatial pattern of signaling all matter. Brief, pulsatile BDNF exposure drives different gene expression programs than sustained exposure. Dendritic versus somatic TrkB activation triggers different downstream cascades. Any compound that reliably upregulates BDNF-TrkB signaling would, in principle, enhance plasticity, but the specific pattern of upregulation determines whether the effect is pro-learning, pro-survival, or both.
Signaling Cascade
BDNF Binds TrkB → Three Pathways Activate Simultaneously
Semax increases both BDNF (the signal) and TrkB (the receptor), amplifying this cascade at two levels
Semax → BDNF protein ↑1.4x · BDNF mRNA ↑3x · TrkB phosphorylation ↑1.6x
Dolotov et al. (2006), rat hippocampus, single dose
Drives long-term potentiation (LTP) — the cellular basis of learning. Activates CREB, which turns on genes for synaptic proteins and more BDNF.
Stronger connections between neurons
Blocks pro-death proteins (BAD, caspase-9) and protects mitochondria. Most critical during stroke or injury when neurons face energy failure.
Neurons survive insults that would otherwise kill them
Regulates calcium signaling and protein kinase C at synaptic terminals. Controls how much neurotransmitter is released per signal.
Fine-tunes communication strength between neurons
Source: Dolotov et al. (2006), Brain Research; standard BDNF-TrkB signaling literature
 View as image
View as imageHow Semax Increases BDNF: The Core Evidence
Hippocampal BDNF and TrkB Upregulation
The landmark study on Semax and BDNF came from Dolotov et al. in 2006. Using adult male rats, they measured BDNF protein levels, TrkB phosphorylation, and mRNA expression in the hippocampus after a single intraperitoneal injection of Semax.[1]
The results were specific and quantifiable. BDNF protein increased 1.4-fold above baseline. TrkB tyrosine phosphorylation (the measure of receptor activation) increased 1.6-fold. At the mRNA level, exon III BDNF transcript levels rose 3-fold, and TrkB mRNA rose 2-fold. These changes occurred simultaneously, meaning Semax increased both the ligand and its receptor, amplifying the signal at two levels of the pathway.
The study also demonstrated selectivity. Semax did not alter the expression of p75NTR, the low-affinity neurotrophin receptor that can trigger apoptosis. This distinction matters: a compound that increased both TrkB (pro-survival) and p75NTR (pro-death) signaling would produce ambiguous effects. Semax specifically enhanced the pro-plasticity arm of neurotrophin signaling.
Specific Binding in the Basal Forebrain
In a companion study published the same year, Dolotov et al. demonstrated that radiolabeled Semax binds specifically to membranes in the rat basal forebrain and increases BDNF protein levels in this region.[2] The basal forebrain houses the nucleus basalis of Meynert, the primary source of cortical cholinergic innervation. These cholinergic neurons are among the first to degenerate in Alzheimer's disease.
The binding was saturable and displaceable, hallmarks of specific receptor interaction rather than nonspecific membrane association. This finding suggests Semax acts through a defined binding site rather than through generalized membrane effects, though the exact receptor has not been fully characterized.
BDNF Expression Across Multiple Brain Regions
Dolotov et al. had earlier shown, in 2003, that Semax stimulates BDNF expression in multiple brain regions simultaneously, including the hippocampus, cortex, and cerebellum.[8] This broad regional effect distinguishes Semax from interventions that elevate BDNF only locally (such as focal electrical stimulation) and aligns with its systemic cognitive effects.
Temporal Dynamics of Neurotrophin Gene Expression
Shadrina et al. (2010) mapped the time course of BDNF and nerve growth factor (NGF) gene expression after Semax administration in the hippocampus, frontal cortex, and retina.[9]
Gene expression changes began within 20 minutes, but the pattern was region-specific and biphasic. In the hippocampus, expression initially decreased at 20 minutes, returned to baseline by 40 minutes, then increased above baseline by 90 minutes. In the frontal cortex, the initial response was an increase rather than a decrease. The retina showed yet another temporal profile. These region-specific dynamics suggest that Semax does not simply flip a global switch on neurotrophin production. Instead, it modulates expression through region-dependent regulatory mechanisms, possibly reflecting differences in local receptor density, enzymatic processing, or transcription factor availability.
Both BDNF and NGF responded, but with different kinetics. NGF changes preceded BDNF changes in most regions, raising the possibility that NGF upregulation is an upstream event that contributes to subsequent BDNF elevation.
Beyond BDNF: Semax and Monoamine Systems
BDNF upregulation does not occur in isolation. Semax also activates dopaminergic and serotonergic neurotransmission, systems that are themselves regulated by BDNF and that in turn modulate BDNF expression. This creates potential positive feedback loops.
Eremin et al. (2004, 2005) conducted detailed neurochemical analyses of Semax's effects on monoamine systems.[3][10] Their microdialysis studies in freely moving rats showed that Semax alone increased striatal levels of the serotonin metabolite 5-HIAA by 25% within 2 hours, with extracellular 5-HIAA gradually rising to 180% of baseline over 1 to 4 hours. For dopamine, Semax alone did not change baseline levels, but when administered 20 minutes before D-amphetamine, it dramatically potentiated the amphetamine-induced dopamine release and locomotor activation.
This pattern, serotonin modulation at baseline plus dopamine potentiation under challenge, suggests Semax primes monoamine circuits rather than directly flooding them with neurotransmitter. The distinction is pharmacologically meaningful. A compound that directly releases dopamine (like amphetamine) produces tolerance, dependence, and neurotoxicity at high doses. A compound that sensitizes dopaminergic circuits to other inputs has a fundamentally different risk profile.
The relationship between BDNF and monoamines runs in both directions. BDNF promotes the survival, differentiation, and synaptic function of both serotonergic neurons in the raphe nuclei and dopaminergic neurons in the ventral tegmental area and substantia nigra. Serotonin and dopamine, in turn, regulate BDNF expression through their respective receptor signaling pathways. This bidirectional regulation means that Semax's monoamine effects may be downstream consequences of its BDNF upregulation, or they may represent a parallel mechanism that feeds back into BDNF expression, or both. Disentangling these pathways would require selective blockade experiments that have not yet been performed.
For more on how peptides interact with dopamine signaling, see Dopamine and Peptide Modulation: The Chemistry of Wanting.
Neuroprotection: Where BDNF Meets Ischemia
The most clinically advanced application of Semax is in stroke recovery, where it has been used in Russian clinical practice. The neuroprotective mechanism connects directly to BDNF-TrkB signaling, which promotes neuronal survival under ischemic stress.
Experimental Stroke Models
Romanova et al. (2006) demonstrated that Semax has neuroprotective and antiamnesic effects during experimental ischemic infarction of the cerebral cortex in rats.[11] Animals treated with Semax showed reduced infarct volume and preserved memory function compared to untreated controls. For the full evidence on Semax in stroke recovery, see Semax for Stroke Recovery: Neuroprotection Evidence.
Gene Expression Profiling After Ischemia
Medvedeva et al. (2017) used genome-wide RNA-Seq to analyze how Semax affects gene expression during ischemic brain injury.[12] Ischemia activates inflammatory genes and suppresses neurotransmitter genes. Semax reversed this pattern: it suppressed inflammatory gene expression and activated neurotransmitter genes. The immune response was the most prominently affected system, with Semax increasing immune cell mobility and enhancing chemokine and immunoglobulin gene expression in ways that promoted tissue repair rather than tissue damage.
Protein-Level Confirmation
Sudarkina et al. (2021) confirmed these transcriptomic findings at the protein level using a rat model of cerebral ischemia-reperfusion.[13] Brain protein expression profiling confirmed that Semax shifts the proteome toward neuroprotective and neurotrophic protein patterns during ischemic recovery. This multi-omics convergence (mRNA, protein, and functional outcomes all aligning) strengthens the case that Semax's neuroprotective effects are genuine and mechanistically coherent.
Additional Neuroprotective and Neuromodulatory Evidence
Dopaminergic Neuroprotection (Parkinson's Model)
Levitskaya et al. (2004) tested Semax in the MPTP model of Parkinson's disease, which selectively destroys dopaminergic neurons in the substantia nigra.[14] Semax protected against MPTP-induced behavioral and neurochemical deficits, consistent with BDNF's known role in dopaminergic neuron survival. Slominsky et al. (2017) extended this to the 6-OHDA model, showing that both Semax and the related peptide Selank affected behavior in rats with Parkinson's-like symptoms.[4]
Reversing Early-Life Neurochemical Disruption
Glazova et al. (2021) demonstrated that Semax can reverse the lasting neurochemical and behavioral damage caused by neonatal SSRI exposure in rats.[5] Early-life fluvoxamine treatment caused anxiety-like behavior, impaired learning, and altered monoamine levels in adulthood. Semax administration normalized these deficits, reversing both behavioral and neurochemical markers. This finding suggests Semax can modulate developmental neuroplasticity, not just adult plasticity.
Copper Binding and Oxidative Protection
Tabbi et al. (2015) showed that Semax has high affinity for copper(II) ions and protects cells against copper-induced oxidative damage.[6] Copper dysregulation contributes to neurodegeneration in Alzheimer's and Wilson's diseases. Semax's ability to chelate copper while simultaneously upregulating BDNF represents a dual neuroprotective mechanism: reducing oxidative damage at the same time as promoting neurotrophic support.
Spinal Cord Recovery
Liu et al. (2025) identified a new mechanism for Semax in spinal cord injury recovery. Semax targets the mu-opioid receptor gene Oprm1, promoting deubiquitination pathways that enhance functional recovery after spinal cord injury in mice.[15] This opioid receptor mechanism is distinct from the BDNF-TrkB pathway, indicating that Semax engages multiple molecular targets.
ADHD and Rett Syndrome
Tsai (2007) proposed Semax as a potential agent for ADHD and Rett syndrome based on its combined effects on BDNF, dopamine, and serotonin systems.[16] This remains a hypothesis paper without clinical trial data, but it illustrates how the convergence of Semax's molecular effects maps onto conditions characterized by neurotrophic and monoamine dysfunction. For the cognitive performance evidence, see Semax and Memory: Cognitive Performance Research.
Evidence Landscape and Limitations
The Semax-BDNF evidence has genuine strengths. Multiple independent research groups have confirmed BDNF upregulation across different brain regions, time points, and experimental models. The effects are quantified with specific fold-changes and backed by both mRNA and protein-level data. The neuroprotective findings in ischemia models have been validated at transcriptomic, proteomic, and functional levels.
The limitations are also real. Nearly all molecular data comes from rodent studies. The fold-changes (1.4x BDNF protein, 3x mRNA) are moderate, not dramatic. Most studies used intraperitoneal injection rather than the intranasal route used clinically. The specific receptor through which Semax initiates BDNF upregulation remains unidentified, which means the complete signaling cascade is not yet mapped. Clinical outcome data from controlled trials in Western medical systems is scarce; the stroke application is based primarily on Russian clinical use rather than multinational randomized trials meeting FDA or EMA standards.
There is also a publication bias concern. Much of the foundational Semax research comes from a small number of Russian laboratories, with limited independent replication from Western groups. This does not invalidate the findings, but it means the evidence base is narrower than for peptides studied across multiple international research centers.
For the full history of how Semax moved from Soviet laboratory to clinical use, see The History of Semax: From Soviet Research to Modern Nootropic Interest. For a broader overview of what Semax research shows across all applications, see Semax: The Russian Nootropic Peptide and What Research Shows.
How Semax Compares to Other BDNF-Elevating Interventions
| Intervention | BDNF Effect | Route | Evidence Level |
|---|---|---|---|
| Semax | 1.4x protein, 3x mRNA in hippocampus | Intranasal/IP | Preclinical (rodent) |
| Acute aerobic exercise | d = 0.83 effect size on serum BDNF | Systemic | Clinical (meta-analysis) |
| SSRIs (chronic) | Normalize reduced BDNF in depression | Oral | Clinical (multiple RCTs) |
| Ketamine (single dose) | Rapid BDNF increase in prefrontal cortex | IV/intranasal | Clinical (limited) |
| Selank | BDNF modulation in hippocampus/PFC | Intranasal | Preclinical (rodent) |
| Cerebrolysin | Neurotrophic factor cocktail | IV | Clinical (stroke trials) |
The comparison reveals that Semax occupies a specific niche: a targeted peptide intervention with well-characterized molecular effects on BDNF-TrkB signaling, but with clinical validation limited to the Russian medical system. Exercise remains the most robustly validated BDNF intervention across human populations.
The Bottom Line
Semax upregulates BDNF protein and mRNA across multiple brain regions, increases TrkB receptor activation, and modulates dopaminergic and serotonergic neurotransmission. The molecular evidence for neuroplasticity enhancement is consistent across multiple studies, models, and measurement levels. The translational gap between rodent molecular data and validated human clinical outcomes remains the central limitation of the Semax evidence base.
Sources & References
- 1RPEP-01129·Dolotov, Oleg V et al. (2006). “Semax Boosts Brain-Derived Neurotrophic Factor (BDNF) in the Hippocampus.” Brain research.Study breakdown →PubMed →↩
- 2RPEP-01130·Dolotov, Oleg V et al. (2006). “Semax Specifically Binds Brain Cells and Increases BDNF Protein Levels.” Journal of neurochemistry.Study breakdown →PubMed →↩
- 6RPEP-02805·Tabbì, Giovanni et al. (2015). “The Neuropeptide Semax Binds Toxic Copper and Protects Brain Cells From Metal-Induced Damage.” Journal of inorganic biochemistry.Study breakdown →PubMed →↩
- 4RPEP-03474·Slominsky, P A et al. (2017). “Selank Peptide Reduced Anxiety in Rats with Parkinson's-like Brain Damage.” Doklady biological sciences : proceedings of the Academy of Sciences of the USSR.Study breakdown →PubMed →↩
- 5RPEP-05410·Glazova, Nataliya Yu et al. (2021). “Semax Peptide Reverses Brain and Behavioral Damage From Prenatal Antidepressant Exposure in Rats.” Neuropeptides.Study breakdown →PubMed →↩
- 15RPEP-12235·Liu, Rongjie et al. (2025). “Semax Promoted Spinal Cord Injury Recovery in Mice by Targeting Opioid Receptors.” British journal of pharmacology.Study breakdown →PubMed →↩