BDNF: The Brain's Growth Factor for New Connections
Neurotrophic Peptides and Brain Health
12.3 kDa
BDNF is a 12.3 kDa neurotrophic protein purified from pig brain in 1982. It binds TrkB receptors to activate MAPK, PI3K, and PLC-gamma signaling cascades that drive synaptic plasticity, neuronal survival, and adult hippocampal neurogenesis.
Barde et al., EMBO J, 1982
Barde et al., EMBO J, 1982
If you only read one thing
BDNF is a protein your brain makes to grow new connections between neurons. It's essential for memory, mood, and keeping brain cells alive. Low BDNF is linked to depression, Alzheimer's, and Huntington's. The easiest way to raise it is exercise — especially high-intensity aerobic — which reliably boosts BDNF with a large effect. You can't take BDNF as a supplement: it doesn't cross the gut wall when swallowed, can't cross the blood-brain barrier when injected, and is too short-lived in the blood. The practical paths are exercise, antidepressants, and peptides like Semax or GLP-1 agonists that tell your brain to produce its own BDNF.
In 1982, Yves-Alain Barde and Hans Thoenen at the Max Planck Institute purified a new protein from pig brain that kept embryonic chick sensory neurons alive in culture.[1] The purification was heroic: approximately 1 microgram of active protein was extracted from 1.5 kilograms of brain tissue, a 1.4 million-fold purification. The resulting protein had a molecular weight of 12,300 Da, was strongly basic (pI of 10.1 or above), and showed neurotrophic activity comparable to nerve growth factor (NGF) but with different antigenic properties and neuronal specificity. They named it brain-derived neurotrophic factor, or BDNF.
BDNF turned out to be far more than a survival factor. Over the following four decades, it emerged as a central regulator of synaptic plasticity, learning, memory, mood, and adult neurogenesis. Reduced BDNF signaling has been implicated in depression, Alzheimer's disease, Huntington's disease, schizophrenia, and anxiety disorders. Exercise increases BDNF levels. Antidepressants increase BDNF levels. A common genetic variant (Val66Met) that impairs BDNF secretion affects episodic memory and hippocampal volume in humans. BDNF sits at the intersection of neuroscience, psychiatry, and peptide biology.
For how BDNF fits into the broader family of neurotrophic growth factors, see Neurotrophic Peptides: Growth Factors That Keep Your Brain Healthy. For the story of BDNF's predecessor, see Nerve Growth Factor (NGF): The Original Neurotrophic Peptide.
Key Takeaways
- BDNF is a protein your brain makes to grow new connections between neurons — it's essential for memory, mood, and keeping brain cells alive.
- Low BDNF is linked to depression, Alzheimer's, and Huntington's. Antidepressants work in part by raising BDNF over weeks of treatment.
- Exercise is the single most reliable way to raise BDNF. Aerobic exercise produces a large measurable spike in your blood within hours.
- You can't buy BDNF as a supplement. It doesn't survive digestion when swallowed, and can't cross the blood-brain barrier when injected.
- About 1 in 3 people carry a genetic variant (Val66Met) that reduces BDNF release. It measurably affects memory performance and hippocampus size.
- Some peptides work by telling your brain to make its own BDNF. Semax upregulates BDNF in the hippocampus — one proposed mechanism for its cognitive effects.
- BDNF is why the story that "you can't grow new brain cells after childhood" is wrong. Adult neurogenesis exists, and BDNF is one of the main reasons.
What BDNF Does in the Brain
BDNF is the most abundant and widely distributed neurotrophin in the mammalian brain. It is expressed at highest levels in the hippocampus (critical for memory), the cortex (cognition and perception), and the basal forebrain (cholinergic neurons that degenerate in Alzheimer's disease). BDNF acts through three interconnected functions.
Synaptic Plasticity
BDNF is required for long-term potentiation (LTP), the cellular mechanism underlying learning and memory. When a synapse is repeatedly activated, BDNF is released from the postsynaptic neuron and acts on both pre- and postsynaptic compartments. On the presynaptic side, BDNF increases vesicle cycling and neurotransmitter release. On the postsynaptic side, it enhances NMDA receptor trafficking and increases calcium influx, strengthening the synaptic connection. This bidirectional modulation makes BDNF an instructive mediator of plasticity: it does not simply support existing connections but actively shapes which connections grow stronger and which weaken. Mice lacking BDNF in the hippocampus show severe LTP deficits and memory impairment.
Neuronal Survival
BDNF promotes survival of existing neurons by activating anti-apoptotic signaling pathways (PI3K/Akt) and suppressing pro-apoptotic signals. During brain development, BDNF determines which neurons survive the period of naturally occurring programmed cell death. In the adult brain, BDNF continues to protect neurons against excitotoxicity, oxidative stress, and metabolic insults. This neuroprotective function is why reduced BDNF is associated with neurodegenerative disease progression.
Adult Neurogenesis
BDNF promotes the birth, differentiation, and integration of new neurons in the adult hippocampus, one of only two brain regions where neurogenesis continues throughout life. New hippocampal neurons contribute to pattern separation (distinguishing similar memories) and mood regulation. The link between BDNF, neurogenesis, and mood is one mechanism by which exercise and antidepressants may exert their effects.
The TrkB Signaling Cascade
BDNF exerts its effects by binding to tropomyosin receptor kinase B (TrkB), a transmembrane receptor tyrosine kinase. When BDNF binds, TrkB dimerizes and autophosphorylates tyrosine residues in its intracellular kinase domain, initiating three major signaling cascades.
MAPK/ERK pathway. Phosphorylated TrkB recruits Shc and Grb2 adapter proteins, activating Ras, Raf, MEK, and ERK (extracellular signal-regulated kinase). ERK translocates to the nucleus, where it phosphorylates the transcription factor CREB (cAMP response element-binding protein). Activated CREB drives expression of genes involved in synaptic plasticity, including Arc, Homer1, and BDNF itself (creating a positive feedback loop). This pathway is the primary mediator of BDNF's effects on learning and memory.
PI3K/Akt pathway. TrkB activates phosphoinositide 3-kinase (PI3K), which generates PIP3 and activates Akt (protein kinase B). Akt phosphorylates and inactivates pro-apoptotic proteins (Bad, caspase-9), promotes mTOR-dependent protein synthesis at synapses, and activates NF-kappa-B-mediated survival gene expression. This pathway mediates BDNF's neuroprotective and cell survival functions.
PLC-gamma/PKC pathway. TrkB phosphorylates phospholipase C-gamma (PLC-gamma), which cleaves PIP2 into IP3 and DAG. IP3 releases calcium from intracellular stores, while DAG activates protein kinase C (PKC). This calcium/PKC signaling modulates neurotransmitter release, ion channel activity, and synaptic vesicle cycling. It is the primary pathway through which BDNF enhances neurotransmission on rapid timescales.
BDNF also binds the low-affinity p75NTR receptor, which can paradoxically promote apoptosis under certain conditions. The balance between TrkB (survival) and p75NTR (death) signaling determines the net effect of BDNF on a given neuron. ProBDNF, the unprocessed precursor form, preferentially binds p75NTR and promotes synaptic weakening (long-term depression), while mature BDNF preferentially binds TrkB and promotes synaptic strengthening (LTP). This yin-yang between proBDNF and mature BDNF adds a layer of regulatory complexity.
The Val66Met Polymorphism
Approximately 20-30% of humans (varying by ethnicity; higher in Asian populations) carry the Met allele of a common single nucleotide polymorphism (rs6265) in the BDNF gene. This Val66Met substitution does not affect the mature BDNF protein itself but impairs the intracellular trafficking and activity-dependent secretion of BDNF from neurons.
Egan et al. demonstrated the functional consequences in a landmark 2003 study.[2] Neurons transfected with Met-BDNF showed lower depolarization-induced secretion, while constitutive (baseline) secretion was unchanged. Met-BDNF failed to localize to secretory granules or synapses. In human subjects, Met carriers showed poorer episodic memory, abnormal hippocampal activation on fMRI during memory tasks, and lower hippocampal N-acetyl aspartate (a marker of neuronal integrity). The interaction between genotype and hippocampal response during memory encoding accounted for 25% of total variation in recognition memory performance.
The Val66Met finding has several implications. It demonstrates that activity-dependent BDNF secretion, not just total BDNF levels, is critical for memory function. It provides a genetic explanation for some of the normal variation in human memory ability. And it suggests that therapeutic strategies targeting BDNF must enhance regulated secretion, not just production.
BDNF, Exercise, and Depression
The relationship between BDNF, exercise, and depression represents one of the most clinically relevant stories in neurotrophin biology.
The Depression Connection
Patients with major depressive disorder consistently show lower serum BDNF levels compared to healthy controls. Post-mortem studies find reduced BDNF expression in the hippocampus and prefrontal cortex of suicide victims. Nearly all effective antidepressant treatments, including SSRIs, SNRIs, tricyclics, MAOIs, ketamine, and electroconvulsive therapy, increase BDNF expression in animal models. The "neurotrophin hypothesis of depression" proposes that stress-induced BDNF reduction in the hippocampus contributes to depression, and that antidepressants work partly by restoring BDNF signaling.
This hypothesis has substantial correlational support but important caveats. BDNF reduction may be a consequence of depression rather than a cause. Peripheral serum BDNF does not necessarily reflect brain BDNF levels. And some effective antidepressant interventions do not consistently increase peripheral BDNF. The hypothesis is useful as a framework but should not be treated as established causation.
The Exercise Effect
Exercise is the most reliable non-pharmacological intervention for increasing BDNF. A systematic review of over 100 manuscripts spanning 20 years confirmed that acute exercise elevates serum BDNF with a large effect size (d = 0.83, p < 0.001).[3] High-intensity aerobic exercise produces the largest acute BDNF increases. Resistance exercise, yoga, and mindfulness practices also elevate BDNF, though typically with smaller effect sizes.
In clinical populations, exercise-induced BDNF increases correlate with depression improvement. One study found that plasma BDNF change predicted 50% of the variance in depression score improvement (R-squared = 0.50, p = 0.002) and 38% of sleep quality improvement (R-squared = 0.38, p = 0.011). In that study, 75% of patients who exercised showed either therapeutic response or complete remission versus 25% of non-exercisers.[3]
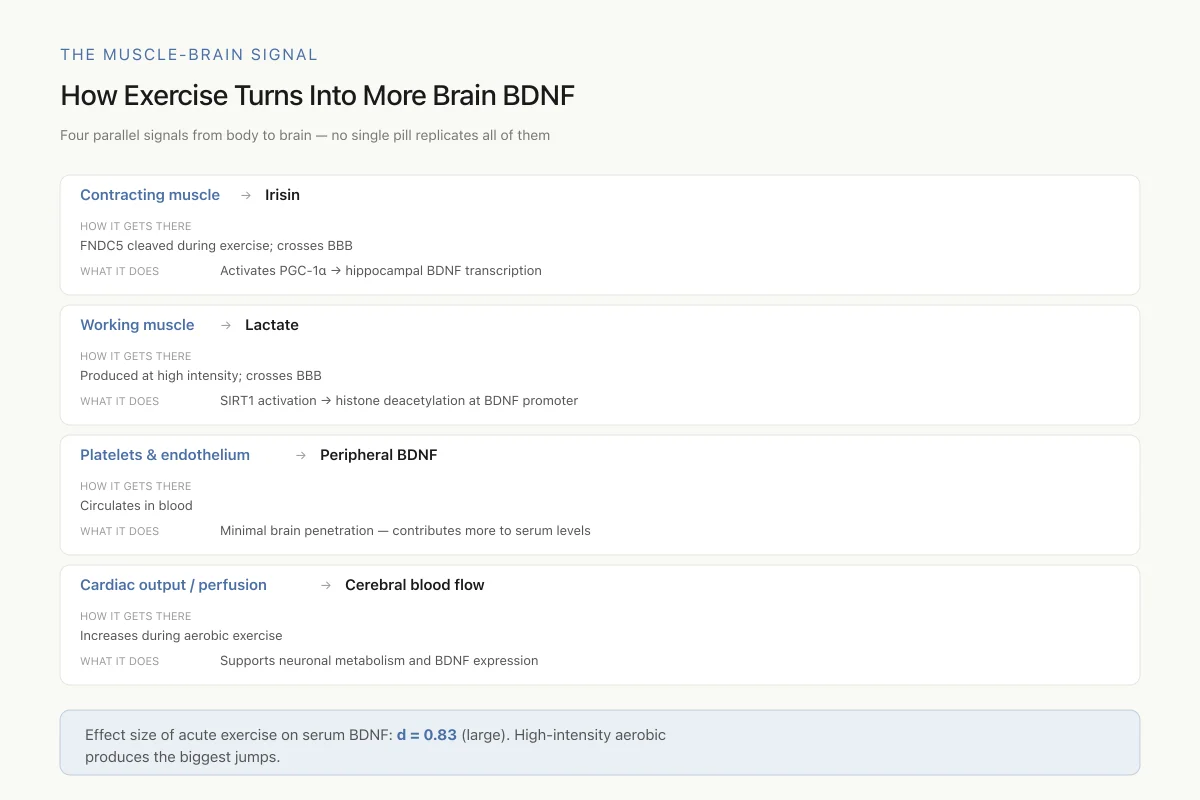
The Muscle-Brain Signal
How Exercise Turns Into More Brain BDNF
Four parallel signals from body to brain — no single pill replicates all of them
Effect size of acute exercise on serum BDNF: d = 0.83 (large). High-intensity aerobic produces the biggest jumps. The reason no pharmacological BDNF booster matches exercise is that exercise hits multiple signals at once — irisin, lactate, blood flow, and more.
Source: Marinus et al. (2023), Frontiers in Physiology — 100+ manuscript review
 View as image
View as imageThe mechanism connecting exercise to brain BDNF involves multiple pathways. Contracting muscles release myokines, including irisin (cleaved from the membrane protein FNDC5), which crosses the blood-brain barrier and stimulates hippocampal BDNF expression through PGC-1-alpha signaling. Exercise-generated lactate also crosses the blood-brain barrier and promotes BDNF transcription through SIRT1 activation and histone deacetylation at the BDNF promoter. Peripheral BDNF from platelets and endothelial cells contributes to circulating levels but may not significantly penetrate the brain. The convergence of these multiple exercise-induced signals on BDNF expression in the hippocampus helps explain why exercise is the most reliable intervention for increasing brain BDNF and why no single pharmacological agent fully replicates the exercise effect.
Peptides That Modulate BDNF
Several peptides in the research literature increase BDNF expression or activity, providing molecular mechanisms for their cognitive and neuroprotective effects. For a comprehensive look at how peptides modulate brain plasticity, see How Peptides Can Modulate Neuroplasticity: Mechanisms and Evidence.
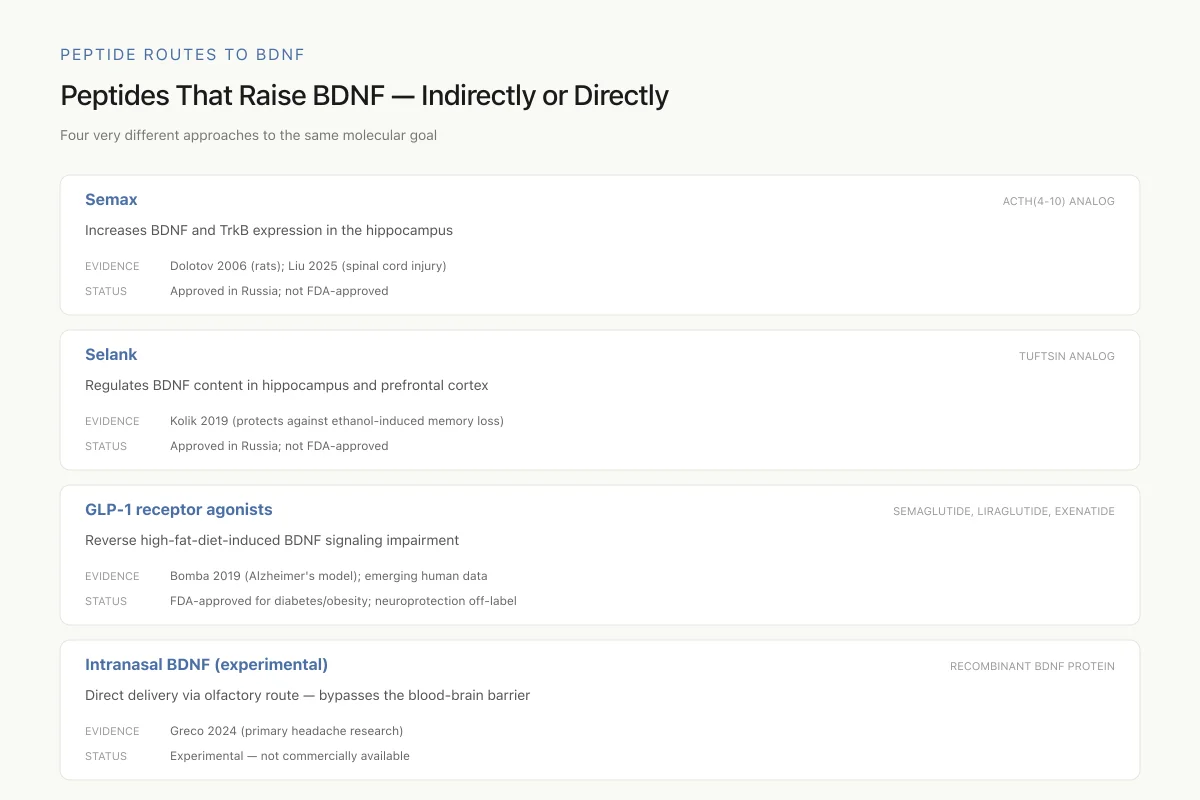
Peptide Routes to BDNF
Peptides That Raise BDNF — Indirectly or Directly
Four very different approaches to the same molecular goal
Semax and Selank are approved and used in Russia but remain in research status in the US. GLP-1 agonists are FDA-approved for other indications and their BDNF effects are emerging as a secondary benefit. Direct intranasal BDNF remains experimental.
Source: Dolotov 2006; Kolik 2019; Bomba 2019; Greco 2024
 View as image
View as imageSemax (ACTH 4-10 Analog)
Semax is a synthetic heptapeptide analog of the ACTH(4-10) fragment, developed in Russia as a nootropic and neuroprotective agent. Dolotov et al. demonstrated that Semax increases both BDNF and TrkB receptor expression in the rat hippocampus, providing a direct molecular link between Semax administration and the neurotrophin system.[4] This upregulation may explain Semax's observed cognitive-enhancing and neuroprotective effects in animal models.
Glazova et al. showed that Semax attenuated behavioral and neurochemical alterations following early-life SSRI exposure in rats, reversing anxiety, learning deficits, and neurotransmitter imbalances.[5] Liu et al. demonstrated that Semax promotes functional recovery after spinal cord injury in mice through a mechanism involving the mu-opioid receptor gene Oprm1 and deubiquitination pathways.[6]
Selank (Tuftsin Analog)
Selank is a synthetic peptide analog of the immunomodulatory peptide tuftsin. Kolik et al. demonstrated that Selank protects against ethanol-induced memory impairment by regulating BDNF content in the hippocampus and prefrontal cortex of rats.[7] This BDNF modulation provides a mechanistic basis for Selank's anxiolytic and nootropic properties.
GLP-1 Receptor Agonists
The diabetes drug class of GLP-1 receptor agonists has shown neuroprotective effects partly mediated through BDNF. Bomba et al. demonstrated that exenatide reverses the high-fat-diet-induced impairment of BDNF signaling and inflammatory response in an animal model of Alzheimer's disease.[8] This finding connects metabolic peptide signaling to neurotrophic pathways and suggests that GLP-1 agonists may have neuroprotective applications beyond diabetes treatment.
Intranasal BDNF Delivery
Greco et al. explored the direct delivery of recombinant human BDNF via intranasal administration as a potential therapy for primary headaches, demonstrating that BDNF itself can be delivered as a therapeutic peptide through a non-invasive route.[9] Intranasal delivery bypasses the blood-brain barrier by exploiting olfactory and trigeminal nerve pathways, offering a potential route for direct neurotrophic peptide therapy.
BDNF in Neurodegeneration
Alzheimer's Disease
BDNF levels decline in the hippocampus and cortex early in Alzheimer's disease, before clinical symptoms appear. Reduced BDNF may contribute to synaptic loss, the strongest pathological correlate of cognitive decline in Alzheimer's. Tayran et al. demonstrated that ABCA7-dependent induction of neuropeptide Y is required for synaptic resilience in Alzheimer's disease through BDNF/NGFR signaling, revealing a peptide-mediated protective pathway.[10] The relationship between BDNF loss and amyloid-beta accumulation remains an active area of investigation. For the amyloid side of the Alzheimer's story, see Amyloid-Beta: The Peptide Fragment at the Heart of Alzheimer's.
Kochanowski et al. measured plasma BDNF alongside activity-dependent neurotrophin protein (ADNP) and vasoactive intestinal peptide (VIP) in treatment-naive patients with multiple sclerosis, demonstrating that neurotrophin profiling with multiple peptide biomarkers may have diagnostic value in neurological disease.[11]
Huntington's Disease
BDNF transport from the cortex to the striatum is impaired in Huntington's disease due to mutant huntingtin protein disrupting BDNF vesicle trafficking along axonal microtubules. Wild-type huntingtin normally promotes BDNF transcription and facilitates its axonal transport from cortical neurons to striatal targets. The polyglutamine expansion in mutant huntingtin disrupts both functions: BDNF gene transcription is reduced (because mutant huntingtin cannot sequester the REST/NRSF transcriptional repressor in the cytoplasm, allowing it to enter the nucleus and silence the BDNF promoter), and the vesicular transport of BDNF along microtubules is impaired. The resulting BDNF deficit in the striatum contributes to the progressive death of medium spiny neurons that characterizes the disease. This makes BDNF delivery to the striatum a therapeutic target, though the blood-brain barrier remains a major obstacle.
Parkinson's Disease
BDNF supports the survival of dopaminergic neurons in the substantia nigra, the population that degenerates in Parkinson's disease. Reduced BDNF expression has been documented in the substantia nigra of Parkinson's patients. Slominsky et al. demonstrated that peptides Semax and Selank affect behavior in rats with 6-OHDA-induced parkinsonism, a finding consistent with their BDNF-modulating properties.[12] Whether BDNF-enhancing strategies can slow dopaminergic neuron loss in humans remains an open question, but the preclinical rationale is strong.
BDNF as a Biomarker
Peripheral BDNF measurement (from serum or plasma) is increasingly used as a biomarker in clinical research. Serum BDNF has been proposed as a biomarker for depression treatment response, cognitive decline in aging, and neurodegeneration progression. The practical challenge is that peripheral BDNF is stored in platelets and released during clotting, making serum BDNF levels dependent on sample handling. Plasma BDNF (which avoids platelet activation) provides a more accurate reflection of circulating BDNF but at much lower concentrations. Standardization of sampling protocols remains an issue for clinical biomarker applications.
Therapeutic Challenges
Despite its central role in brain health, translating BDNF biology into therapy has been difficult.
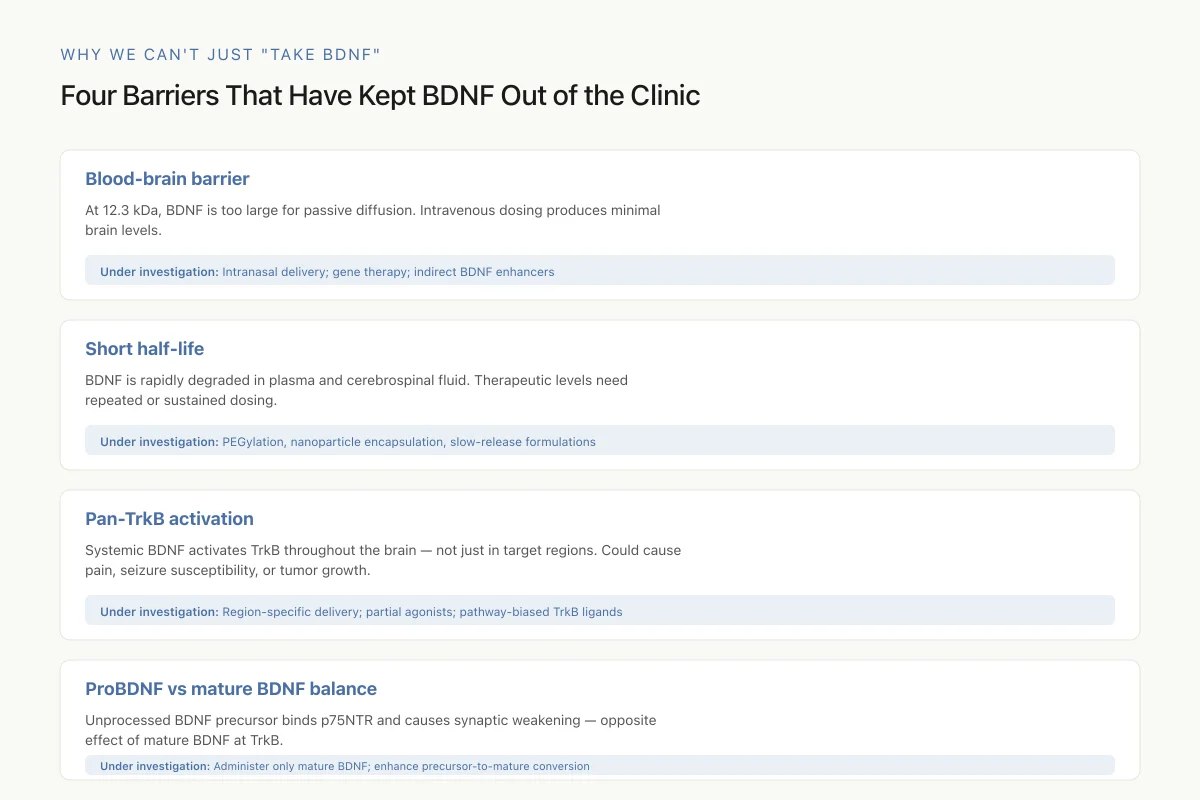
Why We Can't Just "Take BDNF"
Four Barriers That Have Kept BDNF Out of the Clinic
This is why peptides that boost *endogenous* BDNF production (Semax, Selank, GLP-1 agonists) and behavioral interventions (exercise, sleep, antidepressants) are the practical path forward. Direct BDNF administration has fundamental problems that peptide-based BDNF enhancers sidestep.
Source: General BDNF pharmacology; Greco et al. (2024) intranasal delivery research
 View as image
View as imageThe blood-brain barrier blocks peripheral BDNF. Intravenous BDNF does not cross into the brain in meaningful quantities. At 12.3 kDa, BDNF is too large for passive diffusion across the endothelial tight junctions. Intranasal delivery (exploiting olfactory nerve transport) and gene therapy approaches are being explored but remain experimental.
BDNF has a short half-life. The protein is rapidly degraded in plasma and cerebrospinal fluid. Sustained therapeutic levels would require repeated dosing or controlled-release formulations.
Pan-TrkB activation raises safety concerns. BDNF activates TrkB throughout the brain, not just in target regions. Broad TrkB activation could promote unwanted effects, including pain sensitization (TrkB mediates nociceptive signaling), seizure susceptibility (excessive BDNF promotes epileptiform activity), and potentially tumor growth (some cancers express TrkB). Achieving therapeutic benefit requires regional specificity that systemic BDNF delivery cannot provide.
Indirect approaches may be more practical. Exercise, antidepressants, and peptides like Semax increase endogenous BDNF production through physiological regulatory mechanisms, potentially achieving the benefits of BDNF enhancement without the risks of direct protein administration. For how peptide approaches to neuroplasticity are developing, see Neurogenesis and Peptides: Can New Brain Cells Be Stimulated?.
Where BDNF Research Stands
BDNF is one of the most studied molecules in neuroscience, with over 40,000 publications since its discovery. The biology is well established: BDNF drives synaptic plasticity through TrkB signaling, is required for hippocampal memory, declines in depression and neurodegeneration, and increases with exercise and antidepressant treatment.
The therapeutic translation is less advanced. No BDNF-based drug has been approved for any indication. The delivery problem (blood-brain barrier), the specificity problem (pan-TrkB activation), and the stability problem (rapid degradation) have limited direct BDNF therapy. The most promising clinical applications use BDNF as a biomarker (monitoring depression treatment response or neurodegeneration progression) rather than as a therapeutic.
The peptide connection is growing. Semax, Selank, and GLP-1 agonists all modulate BDNF through distinct mechanisms. As the peptide therapeutic field matures, BDNF-enhancing peptides may offer a more practical path to neurotrophic therapy than direct BDNF administration, achieving the biological goal of enhanced neuroplasticity through safer, more targetable molecular tools.
The story that began with 1 microgram of protein from 1.5 kilograms of pig brain has reshaped our understanding of how the brain maintains itself, how it breaks down in disease, and how it might be repaired.
The Bottom Line
BDNF is a 12.3 kDa neurotrophic protein that drives synaptic plasticity, neuronal survival, and adult neurogenesis through TrkB receptor signaling via MAPK/ERK, PI3K/Akt, and PLC-gamma pathways. Reduced BDNF is implicated in depression, Alzheimer's, and Huntington's disease. The Val66Met polymorphism impairs activity-dependent BDNF secretion and accounts for 25% of variation in recognition memory. Exercise increases BDNF with a large effect size (d = 0.83), and peptides like Semax and Selank upregulate BDNF expression in the hippocampus. Direct BDNF therapy remains limited by blood-brain barrier penetration, short half-life, and specificity concerns, making indirect BDNF enhancement through peptides, exercise, and pharmacotherapy the most practical clinical approach.
Sources & References
- 4RPEP-01129·Dolotov, Oleg V et al. (2006). “Semax Boosts Brain-Derived Neurotrophic Factor (BDNF) in the Hippocampus.” Brain research.Study breakdown →PubMed →↩
- 5RPEP-05410·Glazova, Nataliya Yu et al. (2021). “Semax Peptide Reverses Brain and Behavioral Damage From Prenatal Antidepressant Exposure in Rats.” Neuropeptides.Study breakdown →PubMed →↩
- 6RPEP-12235·Liu, Rongjie et al. (2025). “Semax Promoted Spinal Cord Injury Recovery in Mice by Targeting Opioid Receptors.” British journal of pharmacology.Study breakdown →PubMed →↩
- 7RPEP-04287·Kolik, L G et al. (2019). “Selank Peptide Prevented Alcohol-Induced Memory Loss by Regulating BDNF in Rat Brains.” Bulletin of experimental biology and medicine.Study breakdown →PubMed →↩
- 8RPEP-04092·Bomba, Manuela et al. (2019). “Exenatide Reverts the High-Fat-Diet-Induced Impairment of BDNF Signaling and Inflammatory Response in an Animal Model of Alzheimer's Disease..” Journal of Alzheimer's disease : JAD.Study breakdown →PubMed →↩
- 9RPEP-08293·Greco, Rosaria et al. (2024). “Intranasal administration of recombinant human BDNF as a potential therapy for some primary headaches..” The journal of headache and pain.Study breakdown →PubMed →↩
- 10RPEP-09372·Tayran, Hüseyin et al. (2024). “Neuropeptide Y Protects Brain Connections in Alzheimer's Disease Through the ABCA7 Gene.” Cell genomics.Study breakdown →PubMed →↩
- 11RPEP-02689·Kochanowski, Jan et al. (2015). “Neurotrophic Peptides Including VIP Show Normal Levels in Newly Diagnosed Multiple Sclerosis.” Neuro endocrinology letters.Study breakdown →PubMed →↩
- 12RPEP-03474·Slominsky, P A et al. (2017). “Selank Peptide Reduced Anxiety in Rats with Parkinson's-like Brain Damage.” Doklady biological sciences : proceedings of the Academy of Sciences of the USSR.Study breakdown →PubMed →↩