Ipamorelin: The Selective GH Secretagogue
Ipamorelin and GH Secretagogues
200× dose without cortisol rise
Ipamorelin did not raise ACTH or cortisol even at doses more than 200-fold higher than the ED50 for GH release, a selectivity profile unmatched by any other GHRP.
Raun et al., European Journal of Endocrinology, 1998
Raun et al., European Journal of Endocrinology, 1998
If you only read one thing
Ipamorelin is a lab-made peptide that tells your pituitary gland to release growth hormone. What makes it special is what it doesn't do: unlike other peptides in its class, it doesn't raise cortisol (your stress hormone) or prolactin, even at doses 200 times higher than needed. That's why it's called the 'cleanest' growth hormone peptide. The animal science is solid, but no large human trials have been completed, it's not FDA-approved, and the company that developed it walked away from it in the early 2000s.
Ipamorelin is a synthetic pentapeptide that stimulates growth hormone (GH) release from the pituitary gland through the growth hormone secretagogue receptor (GHS-R1a), the same receptor activated by the stomach hormone ghrelin. What distinguishes ipamorelin from every other growth hormone releasing peptide (GHRP) is its selectivity: it releases GH without stimulating cortisol, ACTH, or prolactin, even at doses more than 200 times the amount needed for maximal GH release.[1]
This selectivity, first documented by Raun and colleagues in the landmark 1998 paper that called ipamorelin "the first selective growth hormone secretagogue," remains its defining characteristic in the research literature. This pillar article covers ipamorelin's pharmacology, the evidence base, and its position within the broader GH secretagogue class. For specific comparisons and combinations, see the dedicated articles on ipamorelin vs. other GHRPs, the CJC-1295/ipamorelin combination, and why ipamorelin is considered the "cleanest" GHRP.
Key Takeaways
- What makes ipamorelin different isn't what it does — it's what it skips.
- Even at 200 times the dose needed for growth hormone release, it didn't raise cortisol or prolactin.
- Every other peptide in this class spikes stress hormones or causes ravenous hunger — ipamorelin is the outlier.
- Ipamorelin lasts about two hours in the bloodstream and requires a subcutaneous injection.
- It's called "the cleanest" growth hormone peptide, but no large human trials have ever been completed.
- Novo Nordisk, the company that developed it, walked away in the early 2000s and never filed for approval.
- It pairs so well with CJC-1295 because each peptide flips a different pituitary switch at the same time.
What Is Ipamorelin? Structure and Classification
Ipamorelin (Aib-His-D-2-Nal-D-Phe-Lys-NH2) is a pentapeptide growth hormone secretagogue developed by Novo Nordisk in the 1990s. Its structure was derived from growth hormone releasing peptide-1 (GHRP-1) by removing the central Ala-Trp dipeptide and introducing aminoisobutyric acid (Aib) at the N-terminus for metabolic stability.
The growth hormone secretagogue class includes peptide compounds (GHRP-6, GHRP-2, hexarelin, ipamorelin) and non-peptide compounds (MK-677/ibutamoren). All act through the GHS-R1a receptor on pituitary somatotroph cells to trigger GH release through a pathway distinct from growth hormone-releasing hormone (GHRH).[7] The discovery of this receptor class preceded the identification of ghrelin, its endogenous ligand, by several years. When ghrelin was identified in 1999, the GHS-R1a was renamed the ghrelin receptor.
Ipamorelin is not approved by the FDA for any indication. It exists in the research peptide space and has been used in preclinical and early clinical studies. No large-scale phase 3 trials have been completed. This article covers what the existing research shows and where the evidence gaps are.
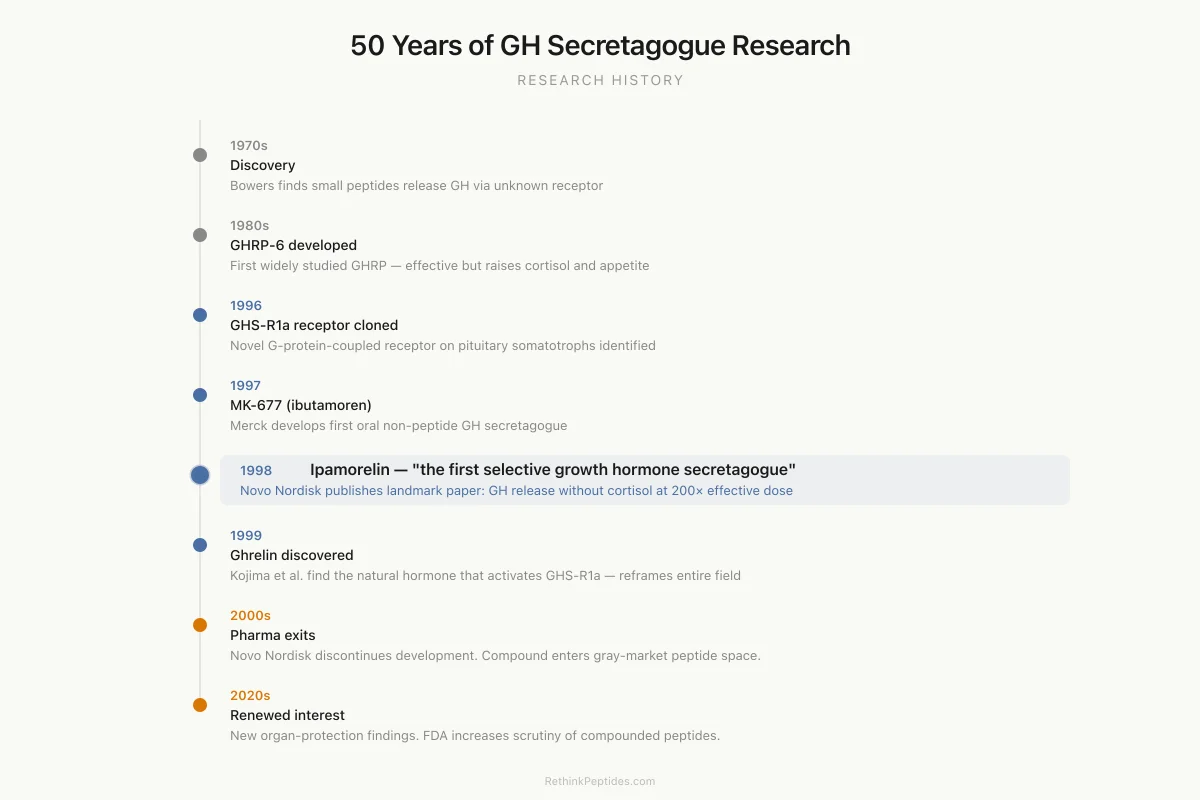
Research History
50 Years of GH Secretagogue Research
Cyril Bowers finds small peptides can release GH from pituitary cells via unknown receptor
First widely studied growth hormone releasing peptide — effective but raises cortisol and appetite
GHS-R1a receptor identified as novel G-protein-coupled receptor on pituitary somatotroph cells
Merck develops oral non-peptide GH secretagogue acting through same receptor
Novo Nordisk publishes landmark paper: "the first selective growth hormone secretagogue"
Kojima et al. discover ghrelin, the natural hormone that activates the same receptor
Novo Nordisk discontinues ipamorelin development. Compound enters gray-market peptide space.
New organ-protection findings (kidney, heart). FDA increases scrutiny of compounded peptides.
Source: Smith (2005); Raun et al. (1998); Kojima et al. (1999)
 View as image
View as imageA Brief History of GH Secretagogues
The GH secretagogue story begins in the 1970s when Cyril Bowers discovered that small synthetic peptides based on enkephalin could release GH from pituitary cells without acting through opioid receptors. This led to the development of GHRP-6, the first widely studied growth hormone releasing peptide, followed by GHRP-2 (more potent) and hexarelin (most potent for GH release).
The puzzle was that all these peptides worked through a receptor that had no known natural ligand. In 1996, the receptor was cloned and characterized as a novel G-protein-coupled receptor on pituitary somatotroph cells.[7] In parallel, Merck developed MK-677 (ibutamoren), a non-peptide oral GH secretagogue acting through the same receptor, demonstrating that small molecules could also activate this pathway.
The endogenous ligand was finally identified in 1999 by Kojima and colleagues: ghrelin, a 28-amino-acid peptide produced mainly by the stomach. This discovery reframed the entire field. The GHS receptor was renamed the ghrelin receptor (GHS-R1a), and the synthetic GH secretagogues were understood to be mimicking a gut-brain signaling axis that linked nutritional status to growth hormone release.
Ipamorelin arrived in 1998, near the peak of GH secretagogue research. Its selectivity profile suggested the possibility of separating GH release from the cortisol and appetite effects that complicated other GHRPs. But the field shifted toward understanding ghrelin biology, and pharmaceutical investment in GH secretagogues for anti-aging applications declined as regulatory scrutiny of growth hormone-related products increased through the 2000s and 2010s.
Mechanism of Action: How Ipamorelin Releases GH
Ipamorelin binds to the GHS-R1a receptor on anterior pituitary somatotroph cells. Receptor activation triggers intracellular calcium release through phospholipase C and inositol trisphosphate (IP3) signaling, which causes GH-containing secretory granules to fuse with the cell membrane and release their contents into the bloodstream.
This pathway is separate from and synergistic with the GHRH pathway. GHRH activates its own receptor (GHRH-R) on the same somatotroph cells, primarily through cAMP-protein kinase A signaling. The two pathways converge on GH release but through different intracellular cascades, which is why combining a GHS-R1a agonist (like ipamorelin) with a GHRH analog (like CJC-1295) produces a synergistic rather than merely additive GH response. This synergy is the rationale behind the CJC-1295/ipamorelin combination.
A 2005 study demonstrated that both peptide and non-peptide GH secretagogues act through the GHS-R1a receptor but can also function as ghrelin receptor ligands with distinct binding properties, suggesting potential differences in downstream signaling despite shared receptor engagement.[6]
The GHS-R1a receptor has constitutive activity, meaning it signals even without a ligand bound. This basal activity suppresses appetite-promoting pathways. Some GHS-R1a agonists increase appetite (GHRP-6 is notorious for this), while others like ipamorelin produce minimal appetite stimulation. The difference likely relates to the specific conformational changes each agonist induces in the receptor, which determines which G-protein pathways are preferentially activated. This concept of "biased agonism" is central to understanding why structurally similar compounds acting on the same receptor can produce different downstream effects.
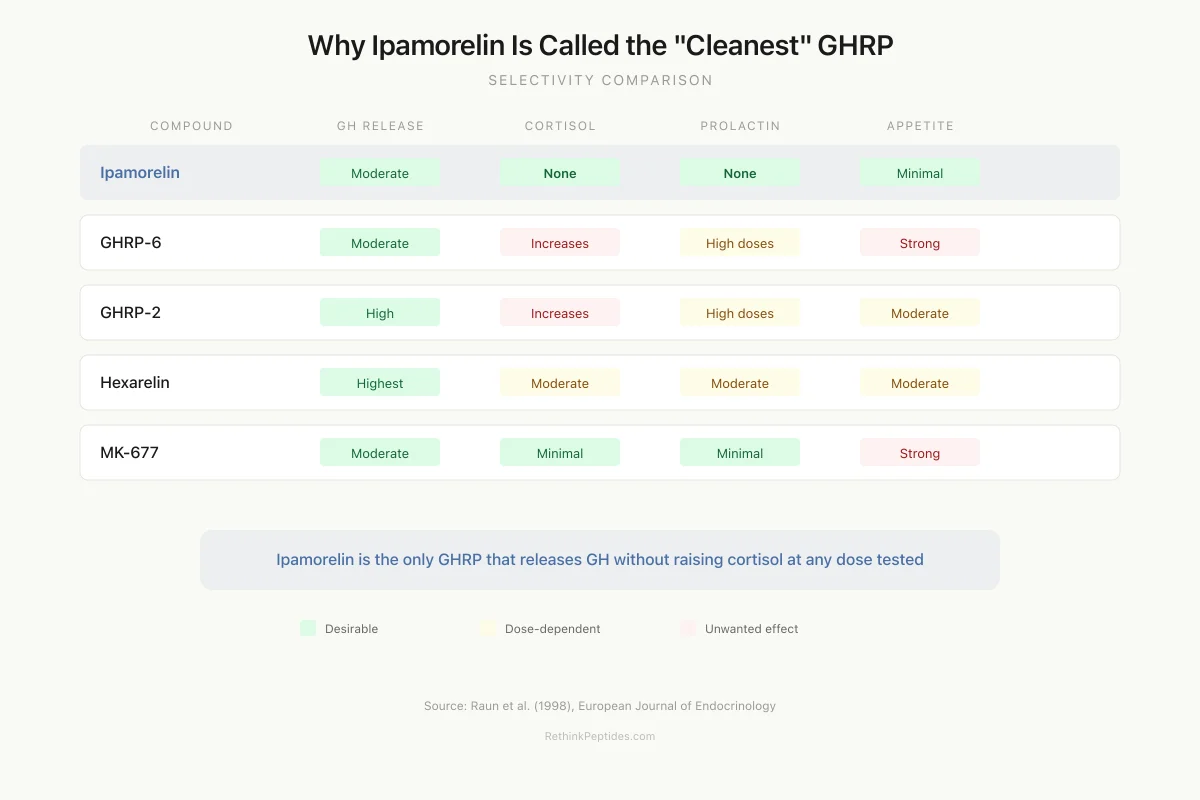
The Selectivity Advantage: Why Ipamorelin Stands Apart
The defining feature of ipamorelin is what it does not do. The landmark 1998 study by Raun et al. compared ipamorelin head-to-head with GHRP-6, GHRP-2, and GHRH in swine.[1]
GH release: All three GHRPs released GH with similar potency. Ipamorelin's ED50 was 2.3 nmol/kg (vs. 3.9 nmol/kg for GHRP-6). Maximum GH levels reached 65 ng/ml for ipamorelin and 74 ng/ml for GHRP-6.
Cortisol and ACTH: GHRP-6 and GHRP-2 both increased plasma cortisol and ACTH at GH-releasing doses. Ipamorelin produced no increase in either hormone at any dose tested, including doses 200-fold above its GH ED50. This selectivity profile matched GHRH, which also does not stimulate the hypothalamic-pituitary-adrenal (HPA) axis.
Prolactin: None of the compounds affected prolactin at the doses tested in the original study, though subsequent work has shown that GHRP-6 and GHRP-2 can raise prolactin at higher doses. Ipamorelin consistently avoids prolactin stimulation.
The mechanism behind this selectivity is not fully resolved. One hypothesis is that GHRP-6 and GHRP-2 have off-target activity at corticotroph cells or hypothalamic CRH neurons that ipamorelin lacks due to its modified structure. Another possibility is that the conformational change induced by ipamorelin at the GHS-R1a receptor produces a biased signaling profile that activates GH release pathways without engaging pathways linked to ACTH secretion. For deeper analysis of these differences, see the article on ipamorelin vs. other GHRPs.
Selectivity Comparison
Why Ipamorelin Is Called the “Cleanest” GHRP
Green = desirable profile. Red = unwanted side effect. Amber = dose-dependent.
Ipamorelin
GHRP-6
GHRP-2
Hexarelin
MK-677
Compound
GH Release
Cortisol
Prolactin
Appetite
Source: Raun et al. (1998), Eur J Endocrinol
 View as image
View as imageHow Ipamorelin Compares to Other GH Secretagogues
| Compound | Type | GH Potency | Cortisol Effect | Prolactin Effect | Half-life | Route |
|---|---|---|---|---|---|---|
| Ipamorelin | Peptide (5 aa) | Moderate | None at any dose | None | ~2 hours | SC injection |
| GHRP-6 | Peptide (6 aa) | Moderate | Increases at GH doses | Increases at high doses | ~20 min | SC injection |
| GHRP-2 | Peptide (6 aa) | High | Increases at GH doses | Increases at high doses | ~25 min | SC injection |
| Hexarelin | Peptide (6 aa) | Highest | Moderate increase | Moderate increase | ~60 min | SC injection |
| MK-677 | Non-peptide | Moderate | Minimal | Minimal | ~5 hours | Oral |
| GHRH (sermorelin) | Peptide (29 aa) | Moderate | None | None | ~12 min | SC injection |
The selectivity pattern is clear: ipamorelin occupies a unique position as the only GHRP with GHRH-like hormonal selectivity while acting through the GHS-R1a receptor rather than the GHRH receptor. This means it can synergize with GHRH analogs (since they activate different receptors) while matching GHRH's clean hormonal profile. For the full comparative analysis, see ipamorelin vs. other GHRPs.
Pharmacokinetics: Absorption, Distribution, Half-Life
Pharmacokinetic studies in multiple species established ipamorelin's absorption and metabolism profile.[2]
After subcutaneous injection, ipamorelin is rapidly absorbed with peak plasma concentrations reached within 15-30 minutes. The elimination half-life is approximately 2 hours, which is relatively short compared to non-peptide GH secretagogues like MK-677 (half-life approximately 5 hours). This short half-life means ipamorelin produces a pulsatile GH release pattern that more closely mimics the body's natural GH secretion rhythm than longer-acting agents.
Ipamorelin is cleared primarily through peptidase degradation and renal excretion. As a pentapeptide, it is subject to the standard limitations of peptide pharmacokinetics: it cannot be taken orally (destroyed by digestive enzymes), and it requires parenteral administration (subcutaneous injection being the most common route in research settings).
Metabolite detection studies have identified specific degradation products of ipamorelin and other GHRPs in urine after nasal and subcutaneous administration, which has enabled anti-doping laboratories to detect their use.[4] These metabolites remain detectable for varying windows depending on the route and dose of administration. GH secretagogues are classified as S2 (Peptide Hormones, Growth Factors, Related Substances and Mimetics) on the WADA Prohibited List and are prohibited at all times, both in and out of competition, in all sports. Athletes testing positive for ipamorelin or its metabolites face standard anti-doping sanctions.
The short half-life of ipamorelin relative to MK-677 has implications for both dosing strategy in research settings and detection windows in anti-doping contexts. MK-677's longer half-life and oral availability make it more convenient but also more detectable. Ipamorelin's rapid clearance creates a narrower detection window but also means its GH-elevating effect is more transient.
Preclinical Evidence: Beyond GH Release
Bone Protection
A 2001 study investigated whether ipamorelin could counteract glucocorticoid-induced bone loss in rats.[3] Glucocorticoids (like prednisolone) are among the most common causes of secondary osteoporosis, and they work partly by suppressing GH/IGF-1 signaling. Ipamorelin treatment preserved bone mineral content and bone mechanical strength in glucocorticoid-treated rats compared to untreated controls. This finding suggested that GH secretagogues might have therapeutic potential for steroid-induced osteoporosis, though this has not been tested in human trials.
A separate study showed that methylprednisolone did not block ipamorelin's GH-releasing effect despite suppressing endogenous GH secretion, demonstrating that the GHS-R1a pathway can override glucocorticoid-mediated GH suppression.[8]
Reproductive Effects
Chronic ipamorelin treatment in young female rats influenced reproductive parameters including follicular development and hormonal cycling, suggesting that GH secretagogues interact with the hypothalamic-pituitary-gonadal axis.[5] GHS-R1a receptors are expressed not only on pituitary somatotrophs but also in the hypothalamic arcuate nucleus, where they are co-localized with neurons involved in reproductive hormone regulation. The effects were modest and the clinical relevance uncertain, but they highlight that GHS-R1a signaling extends beyond the somatotroph axis and has the potential to influence multiple endocrine systems.
Cardioprotection and Organ Protection
GH secretagogues including synthetic GHRPs have demonstrated cardioprotective effects in ischemic heart models.[9] The mechanism appears to involve direct GHS-R1a receptor activation on cardiac cells independent of GH release, since hexarelin (a related GHRP) showed cardioprotective effects even in GH-deficient models.
A 2024 study found that GHS-R1a agonists including ipamorelin and anamorelin inhibited cisplatin-induced kidney injury in preclinical models.[10] Cisplatin is a widely used chemotherapy drug limited by nephrotoxicity. The renal protective effect appeared to be mediated through anti-inflammatory and anti-apoptotic pathways activated by GHS-R1a signaling, independent of the GH-elevating effect. This finding is consistent with the broader observation that GHS-R1a receptors are expressed in multiple tissues beyond the pituitary, including kidney, heart, and immune cells, suggesting pleiotropic protective functions.
Nitrogen Balance and Body Composition
A 2009 study evaluated the effects of GH and GH secretagogues on nitrogen balance and urea synthesis in preclinical models.[12] GH release stimulates protein synthesis and nitrogen retention, which is the rationale for investigating GH secretagogues in catabolic states such as post-surgical recovery, critical illness, prolonged bedrest, and age-related muscle wasting (sarcopenia). The study demonstrated that GH secretagogue-induced GH release could shift nitrogen balance toward anabolism, reducing urea synthesis and promoting protein accretion.
This anabolic effect is the primary reason GH secretagogues have attracted interest in age-related sarcopenia and frailty. As GH secretion declines with age (somatopause), the resulting drop in IGF-1 contributes to progressive loss of lean mass. Restoring pulsatile GH release through GHS-R1a agonism could theoretically counteract this decline. However, the evidence for meaningful clinical benefit in elderly populations remains preliminary, and the risk-benefit calculation must account for potential effects on glucose metabolism and cancer risk.
The AOD-9604 fragment takes a fundamentally different approach by isolating the lipolytic fragment of GH (amino acids 177-191) without its growth-promoting or IGF-1-elevating effects. This represents an alternative strategy for metabolic applications where GH's fat-mobilizing properties are desired without the full GH/IGF-1 axis activation that GH secretagogues produce.
Beyond Growth Hormone
Ipamorelin's Preclinical Effects Across Organs
Preserved bone mineral content and mechanical strength against glucocorticoid-induced loss
Andersen et al. (2001)
Cardioprotective effects in ischemic models, independent of GH release
Frascarelli et al. (2003)
Inhibited cisplatin-induced kidney injury via anti-inflammatory and anti-apoptotic pathways
Lu et al. (2024)
GH release maintained even during glucocorticoid-mediated suppression of endogenous GH
Malmöf et al. (1999)
Shifted nitrogen balance toward anabolism, reduced urea synthesis, promoted protein accretion
Aagaard et al. (2009)
All findings are from animal studies. None have been confirmed in human clinical trials. The organ-protective effects appear to work through GHS-R1a receptors expressed outside the pituitary, independent of GH release itself.
Source: Aggregate preclinical literature, 1999–2024
 View as image
View as imageThe Development of Hybrid Secretagogues
Researchers at Novo Nordisk explored hybrid molecules combining structural elements of ipamorelin with NN703 (another GH secretagogue) to create compounds with enhanced potency while retaining selectivity.[4] These hybrids achieved higher GH release in some assays, demonstrating that ipamorelin's structure could serve as a scaffold for next-generation secretagogue design.
The broader field has moved toward understanding the full pharmacology of the GHS-R1a receptor. A 2021 study identified a putative beta-arrestin "superagonist" of the ghrelin receptor, showing that biased agonism at this receptor could produce functionally distinct outcomes depending on which signaling pathway is preferentially activated.[11] This concept of biased agonism may eventually explain ipamorelin's selectivity profile and guide the design of even more selective GH secretagogues. Understanding which signaling pathways produce GH release versus cortisol release versus appetite stimulation could enable the rational design of compounds that activate only the desired downstream effects. Ipamorelin, despite being discovered through empirical screening rather than rational design, appears to be a natural example of a biased agonist at the GHS-R1a receptor.
Limitations and Evidence Gaps
The ipamorelin research base has substantial gaps:
No completed phase 3 trials. Despite being identified as a promising candidate in 1998, ipamorelin never advanced through full clinical development. Novo Nordisk discontinued its development program, and the compound entered the gray-market peptide space without the large-scale efficacy and safety data that regulatory approval requires.
Short-term studies only. Most studies lasted days to weeks. Long-term effects of chronic GHS-R1a activation on cancer risk (GH and IGF-1 are growth factors), glucose metabolism (GH antagonizes insulin), and joint health are unknown.
Animal data predominates. The selectivity data come primarily from swine and rat models. Human dose-response data for cortisol and prolactin endpoints are extremely limited. Whether the selectivity observed in animals translates perfectly to humans is assumed but not rigorously proven across dose ranges.
Regulatory status. Ipamorelin is not FDA-approved for any indication. It is classified as a research chemical. The FDA has taken enforcement action against compounding pharmacies selling GH secretagogues, and ipamorelin falls within this regulatory scrutiny. It is also a WADA-prohibited substance.
Potential desensitization. GHS-R1a receptors can desensitize with chronic agonist exposure. Whether repeated ipamorelin dosing leads to tachyphylaxis (reduced response over time) has not been systematically studied in long-term protocols.
IGF-1 and cancer risk. Any intervention that chronically elevates GH and IGF-1 carries theoretical cancer risk concerns, since IGF-1 promotes cell proliferation and inhibits apoptosis. This concern applies to all GH secretagogues, not ipamorelin specifically. Epidemiological data linking higher IGF-1 levels within the normal range to modestly increased cancer incidence inform this caution.
Safety
ModerateNo completed clinical development program
Concern
Ipamorelin was abandoned by Novo Nordisk before completing clinical efficacy trials. It entered the gray-market peptide space without the safety and efficacy data that FDA approval requires. Long-term effects of chronic GHS-R1a activation on cancer risk (IGF-1 is a growth factor), glucose metabolism, and joint health are unknown.
What the research says
Short-term animal studies show no cortisol or prolactin effects, and the selectivity profile is genuinely clean. The 2-hour half-life limits sustained IGF-1 elevation compared to longer-acting agents. But 'cleaner than other GHRPs' is not the same as 'proven safe for long-term human use.'
Particularly relevant for: Anyone considering self-administration of ipamorelin
What to do
WADA-prohibited for athletes. Not FDA-approved. Products from gray-market suppliers have no guaranteed purity or potency. Discuss with a healthcare provider, especially regarding IGF-1 and glucose monitoring.
Raun et al. (1998); FDA compounding guidance; WADA Prohibited List
Interaction with GH-affected sleep. GH secretagogues including ipamorelin are sometimes discussed in the context of sleep quality. Endogenous GH release occurs primarily during slow-wave sleep, and GHS-R1a activation can influence sleep architecture. Whether exogenous ipamorelin enhances or disrupts natural sleep-related GH pulsatility depends on timing, dose, and individual factors that have not been systematically studied.
The Bottom Line
Ipamorelin is a pentapeptide GH secretagogue that activates the GHS-R1a (ghrelin) receptor to release growth hormone without stimulating cortisol, ACTH, or prolactin, a selectivity profile documented at doses 200-fold above its effective GH-releasing dose. Preclinical studies show bone-protective, cardioprotective, and nephroprotective effects beyond GH release. The evidence base remains limited to preclinical and small clinical studies; no large phase 3 trials have been completed, and the compound is not approved for any clinical use.
Sources & References
- 1RPEP-00485·Raun, K et al. (1998). “Ipamorelin: The First Growth Hormone Peptide That Only Boosts GH Without Other Hormones.” European journal of endocrinology.Study breakdown →PubMed →↩
- 2RPEP-00466·Johansen, P B et al. (1998). “How Ipamorelin and Other Growth Hormone Peptides Are Absorbed by the Body.” Xenobiotica; the fate of foreign compounds in biological systems.Study breakdown →PubMed →↩
- 3RPEP-00642·Andersen, N B et al. (2001). “Ipamorelin Reverses Steroid-Induced Bone Loss in Adult Rats Over 3 Months.” Growth hormone & IGF research : official journal of the Growth Hormone Research Society and the International IGF Research Society.Study breakdown →PubMed →↩
- 4RPEP-00666·Hansen, T K et al. (2001). “Hybrid GH Secretagogues Combining NN703 and Ipamorelin Features Are Potent In Vitro and In Vivo.” Bioorganic & medicinal chemistry letters.Study breakdown →PubMed →↩
- 5RPEP-00737·Jiménez-Reina, L et al. (2002). “Chronic Ipamorelin Treatment Doesn't Desensitize the GH System in Young Female Rats.” Histology and histopathology.Study breakdown →PubMed →↩
- 6RPEP-01047·Holst, Birgitte et al. (2005). “GH Secretagogues Act as Both Ghrelin Agonists and Allosteric Receptor Modulators.” Molecular endocrinology (Baltimore.Study breakdown →PubMed →↩
- 7RPEP-01088·Smith, Roy G (2005). “The Development of Growth Hormone Secretagogues: From GHRP Discovery to MK-677 and Beyond.” Endocrine reviews.Study breakdown →PubMed →↩
- 8RPEP-00538·Malmlöf, K et al. (1999). “Ipamorelin Still Releases Growth Hormone and Promotes Growth Even During Steroid Treatment.” Growth hormone & IGF research : official journal of the Growth Hormone Research Society and the International IGF Research Society.Study breakdown →PubMed →↩
- 9RPEP-00817·Frascarelli, Sabina et al. (2003). “Ghrelin and Synthetic GH Secretagogues Protect Rat Hearts From Ischemic Damage.” Basic research in cardiology.Study breakdown →PubMed →↩
- 10RPEP-08773·Lu, Zengbing et al. (2024). “Ghrelin-Mimicking Peptides Anamorelin and Ipamorelin Reduce Chemotherapy-Induced Weight Loss — Anamorelin Also Stops Nausea When It Reaches the Brain.” Physiology & behavior.Study breakdown →PubMed →↩
- 11RPEP-05481·Karaki, Fumika et al. (2021). “Discovery of a β-Arrestin Superagonist of the Ghrelin Receptor Opens New Drug Targeting.” ChemMedChem.Study breakdown →PubMed →↩
- 12RPEP-01448·Aagaard, Niels Kristian et al. (2009). “Ipamorelin Improves Nitrogen Balance in Steroid-Treated Rats: Better Than GH Alone.” Growth hormone & IGF research : official journal of the Growth Hormone Research Society and the International IGF Research Society.Study breakdown →PubMed →↩