Peptide Hormone Dysregulation in PCOS
PCOS and Peptide Hormones
6-20% of women affected
Polycystic ovary syndrome affects 6-20% of women of reproductive age, driven by dysregulated peptide hormone signaling across the hypothalamic-pituitary-gonadal axis.
Hoteit et al., Therapeutic Advances in Endocrinology and Metabolism, 2025
Hoteit et al., Therapeutic Advances in Endocrinology and Metabolism, 2025
If you only read one thing
PCOS isn't just an ovary problem — it's a whole-body hormone signaling breakdown. Your brain's hormone clock runs too fast, pumping out too much of one hormone (LH) and not enough of another (FSH). Your ovaries respond by making too much testosterone. Your insulin system is broken too, and high insulin makes the testosterone problem worse. These aren't separate issues — they're feedback loops that amplify each other. GLP-1 drugs (like Ozempic) are the first treatment that hits multiple loops at once, which is why they're showing better results than older approaches.
Polycystic ovary syndrome is not one disease. It is a cascade of peptide hormone dysregulations that amplify each other across the hypothalamic-pituitary-gonadal (HPG) axis, the metabolic system, and the ovary itself. At the hypothalamic level, GnRH neurons fire too fast, driven by overactive kisspeptin signaling and reduced inhibition from dynorphin. At the pituitary, the accelerated GnRH pulse frequency skews gonadotropin secretion toward LH dominance over FSH. At the ovary, excess LH drives androgen production while elevated anti-Mullerian hormone (AMH) blocks follicle maturation. Layered onto this reproductive axis dysfunction is metabolic disruption: hyperinsulinemia amplifies ovarian androgen production while altered incretin dynamics (GLP-1, GIP) compound insulin resistance.[1] Understanding PCOS as a peptide hormone disorder, rather than simply an ovarian condition, explains why GLP-1 receptor agonists are emerging as treatments that address both the metabolic and reproductive dimensions simultaneously.[2] This article maps each peptide pathway that goes wrong in PCOS and how they interconnect. For the specific role of GLP-1 drugs in PCOS treatment, see GLP-1 agonists for PCOS. For the kisspeptin pulse frequency mechanism, see kisspeptin and PCOS.
Key Takeaways
- PCOS affects roughly 1 in 10 women of reproductive age — it's not rare, it's common.
- PCOS isn't really an ovary problem — it's a whole-body hormone signaling breakdown.
- The brain's hormone clock runs nearly twice as fast in PCOS, throwing the whole reproductive system off.
- Birth control hides the symptoms but doesn't actually fix what's wrong — stop the pills and it all returns.
- AMH — a fertility hormone — runs 2 to 3 times higher than normal in women with PCOS.
- GLP-1 drugs like Ozempic are the first treatment that hits both the brain and metabolism at once.
- High insulin makes the testosterone problem worse — fixing blood sugar is part of fixing the hormones.
The GnRH Pulse Generator: Where PCOS Begins
Neuroendocrine Disruption
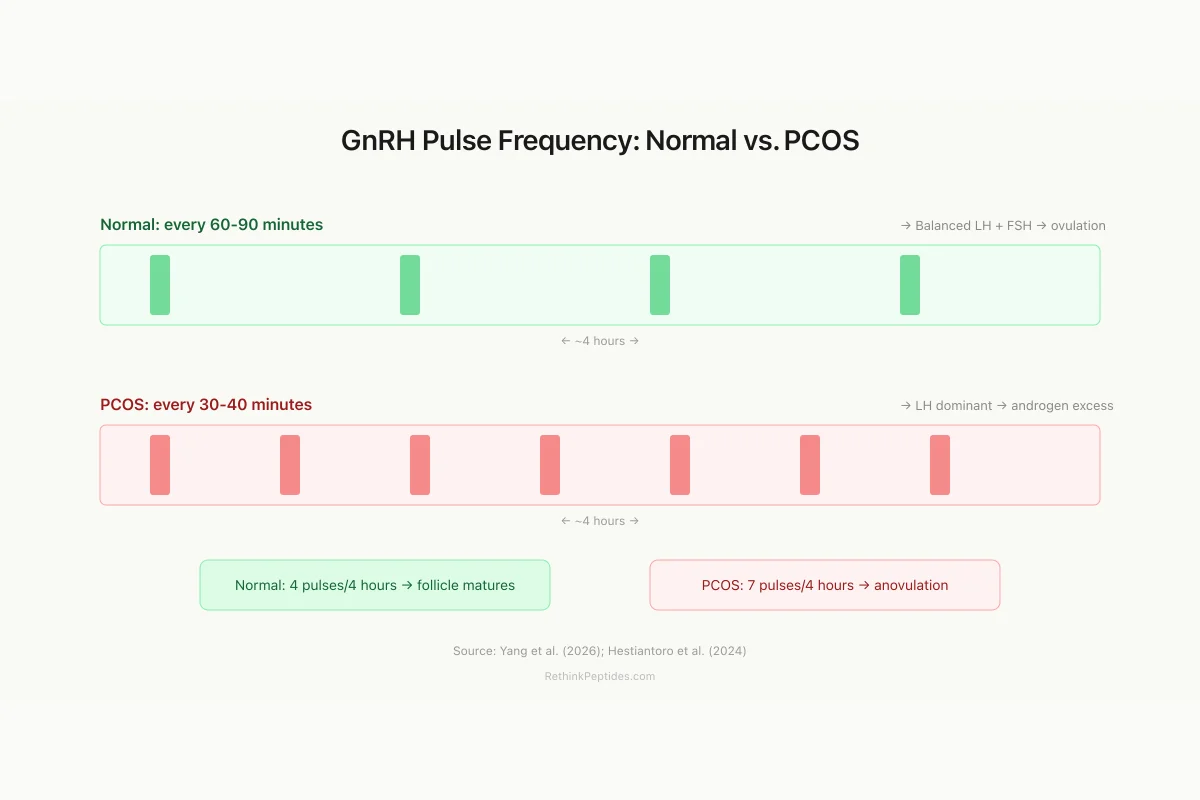
GnRH Pulse Frequency: Normal vs. PCOS
Normal pulse result
Balanced LH:FSH → follicle matures → ovulation
PCOS pulse result
LH excess, FSH suppressed → androgen ↑, no ovulation
Source: Yang et al. (2026); Hestiantoro et al. (2024)
 View as image
View as imageThe hypothalamic GnRH pulse generator sets the rhythm of the entire reproductive axis. In healthy women, GnRH neurons fire in pulses approximately every 60-90 minutes during the follicular phase, a frequency that favors balanced secretion of both LH and FSH from the anterior pituitary. In PCOS, this pulse frequency accelerates, sometimes to every 30-40 minutes. The faster rhythm preferentially drives LH secretion while suppressing FSH, creating the characteristic LH:FSH ratio elevation that defines PCOS neuroendocrinology.
Yang et al. (2026) demonstrated that this GnRH hyperactivity is not merely a secondary consequence of ovarian hormones but a primary driver of PCOS pathology. In a PCOS mouse model, GLP-1 receptor agonist treatment reprogrammed the hypothalamic RASA1/Ras/AKT/GnRH axis, directly normalizing GnRH neuronal activity and rescuing HPG axis function. This central mechanism was distinct from GLP-1's peripheral metabolic effects, proving that the hypothalamic GnRH disruption is a pharmacologically targetable node in PCOS.[3]
The clinical implication is substantial: PCOS treatments that target only peripheral symptoms (androgen excess, anovulation, insulin resistance) may fail because the central pulse generator continues driving the hormonal cascade. Interventions that normalize GnRH pulsatility address the root cause rather than downstream effects.
The GnRH pulse frequency disturbance also explains a clinical paradox: why combined oral contraceptives, which suppress the HPG axis entirely, effectively manage PCOS symptoms during use but provide no lasting benefit after discontinuation. Contraceptives silence the pulse generator without correcting it. When treatment stops, the accelerated pulsatility resumes and symptoms return. An intervention that actually resets the pulse frequency, rather than overriding it, would offer more durable benefit. The Yang et al. finding that GLP-1 agonists can reprogram GnRH axis signaling through the RASA1/Ras/AKT pathway suggests this kind of mechanistic correction may be achievable.
The pulse generator operates as a neuroendocrine oscillator whose frequency depends on the balance of excitatory and inhibitory inputs. Beyond kisspeptin and dynorphin, gamma-aminobutyric acid (GABA), glutamate, and neuropeptide Y all modulate GnRH neuron firing. In PCOS, prenatal androgen exposure may permanently alter the sensitivity of GnRH neurons to these inputs, explaining why the disorder typically manifests at puberty and persists into menopause even as androgen levels decline with age. This developmental programming hypothesis suggests that PCOS begins before birth in women exposed to elevated maternal androgens, setting up the neuroendocrine architecture for accelerated GnRH pulsatility that activates once the HPG axis matures at puberty.
Kisspeptin and KNDy Neurons: The Upstream Switch
GnRH neurons do not generate their own pulse rhythm independently. They are driven by a population of hypothalamic neurons called KNDy neurons, which co-express three peptides: kisspeptin (the accelerator), neurokinin B (the initiator), and dynorphin (the brake). The balance among these three peptides determines GnRH pulse frequency.
Hestiantoro et al. (2024) measured kisspeptin and dynorphin expression in women with PCOS compared to controls. Both peptides were significantly altered: kisspeptin expression was elevated while dynorphin was reduced. This imbalance, more accelerator and less brake, explains the accelerated GnRH pulsatility that characterizes PCOS. The study confirmed at the molecular level what clinical measurements of LH pulsatility had suggested for decades: that PCOS involves a fundamental disruption of the KNDy neuron oscillator.[4]
The kisspeptin elevation in PCOS is not merely a marker of disease. It appears to be a causal driver. Kisspeptin directly activates GnRH neurons through the KISS1R receptor, and elevated kisspeptin signaling increases both the frequency and amplitude of GnRH pulses. This creates a positive feedback loop: excess kisspeptin drives excess GnRH, which drives excess LH, which drives excess ovarian androgen production, and androgens in turn stimulate further kisspeptin expression. Breaking this cycle at the kisspeptin level is an active area of therapeutic research. For the neuroimaging evidence of kisspeptin's role in female arousal, a related but distinct pathway, see kisspeptin: the peptide that triggers puberty and drives desire.
Neurokinin B (NKB), the third member of the KNDy triad, adds another pharmacological target. NKB acts as the "spark plug" of the KNDy oscillator: it initiates each pulse by synchronizing firing among KNDy neurons. In PCOS, NKB signaling is elevated, contributing to the accelerated pulse frequency. NKB receptor antagonists (NK3R antagonists) have been tested in clinical trials for PCOS and show promise in reducing LH levels and testosterone concentrations. The NK3R antagonist approach is conceptually different from GLP-1 therapy: it targets the neuroendocrine oscillator directly rather than addressing the metabolic inputs that drive it. Whether combining NK3R antagonists with GLP-1 agonists would produce additive or synergistic benefits in PCOS has not been tested but has clear pharmacological rationale, as the two approaches intervene at different points in the peptide network.
AMH: The Ovarian Amplifier
Anti-Mullerian hormone (AMH) is produced by granulosa cells of pre-antral and small antral follicles. In healthy women, AMH serves as a gatekeeper that prevents premature follicle recruitment. In PCOS, AMH levels are 2-3 fold higher than normal, reflecting both the increased number of small antral follicles characteristic of polycystic ovaries and an intrinsic increase in AMH production per follicle.
Bhide and Homburg (2016) reviewed the evidence that AMH is not just a biomarker for PCOS but an active participant in its pathophysiology. Elevated AMH inhibits the action of FSH on follicle development, contributing to anovulation. It also reduces aromatase activity in granulosa cells, impairing the conversion of androgens to estrogens and further elevating the hyperandrogenic milieu.[5]
More recent research has added a central dimension to AMH's role. AMH receptors are expressed on GnRH neurons in the hypothalamus, and elevated circulating AMH can directly stimulate GnRH neuron activity. This creates another positive feedback loop: the ovary's excess AMH production feeds back to the brain to accelerate GnRH pulsatility, which in turn drives more LH to the ovary, perpetuating follicle arrest and androgen excess. The AMH-GnRH connection means that PCOS is not a disease where the brain and ovary are independently dysfunctional; rather, they are locked in a self-reinforcing cycle where each organ's peptide hormone output amplifies the other's pathology. For clinical use of AMH as a fertility biomarker, see AMH: the peptide biomarker for fertility.
AMH also has diagnostic utility. Because AMH correlates with antral follicle count and is relatively stable across the menstrual cycle (unlike LH and FSH, which fluctuate substantially), it has become the preferred peptide biomarker for PCOS screening and for monitoring treatment response. Declining AMH levels during GLP-1 agonist treatment correlate with improving ovarian function and return of ovulation, providing a quantitative endpoint for therapeutic efficacy that is more reliable than self-reported menstrual regularity.
Insulin and Incretins: The Metabolic Layer
Insulin resistance is documented in approximately 75% of women with PCOS. Hyperinsulinemia amplifies ovarian androgen production by stimulating theca cell steroidogenesis and by reducing hepatic sex hormone-binding globulin (SHBG) production, which increases the fraction of bioavailable testosterone. This metabolic-reproductive interaction is bidirectional: androgens themselves promote visceral fat deposition and insulin resistance, creating yet another positive feedback loop.
Gunesli et al. (2025) added a new dimension by measuring fasting and postprandial oxytocin and incretin dynamics in women with PCOS. Fasting GLP-1 levels were lower in women with PCOS, and the postprandial GLP-1 response was blunted. GIP dynamics were similarly altered. Oxytocin levels also showed abnormal meal-related patterns. These findings place incretin dysfunction alongside insulin resistance as a metabolic feature of PCOS, not merely a consequence of obesity but a component of the syndrome itself.[6]
Bizon et al. (2025) measured associations between serum GIP, GLP-1, and DPP-4 (the enzyme that degrades incretins) with metabolic and hormonal profiles in PCOS. Their analysis found that incretin levels correlated with metabolic severity independently of BMI, suggesting that incretin dysfunction in PCOS has a hormonal component beyond what can be explained by excess body weight alone.[7]
Bednarz and Javed (2022) reviewed the role of GLP-1 receptor agonists in insulin resistance with concomitant obesity treatment in PCOS, documenting the bidirectional relationship between insulin signaling and reproductive hormone dysfunction. Their review established that insulin resistance in PCOS is not simply a consequence of obesity: lean women with PCOS also show insulin resistance at rates exceeding weight-matched controls. This suggests an intrinsic defect in insulin signaling that is part of the syndrome rather than a secondary metabolic consequence.[8]
The metabolic disruption in PCOS extends to adipose tissue peptide signaling. Adiponectin (an insulin-sensitizing peptide from fat cells) is typically reduced in PCOS, while leptin (the satiety peptide) shows altered dynamics. These adipokine changes interact with the incretin deficits: reduced GLP-1 secretion impairs insulin release, while reduced adiponectin impairs insulin action, creating a compound metabolic deficit that neither peptide abnormality would produce alone. The result is that women with PCOS require substantially higher insulin levels to maintain glucose homeostasis, and this compensatory hyperinsulinemia drives androgen production as a side effect of trying to maintain blood sugar control.
GLP-1 Agonists: Targeting Both Axes
The recognition that PCOS involves both metabolic (insulin, incretin) and reproductive (GnRH, kisspeptin, AMH) peptide dysregulation has made GLP-1 receptor agonists an obvious therapeutic candidate. Unlike metformin, which primarily addresses insulin resistance, GLP-1 agonists act on both the metabolic and central neuroendocrine axes.
Sanchez-Garrido et al. (2024) published in Nature Communications a comparison of GLP-1-based multi-agonists (targeting GLP-1, GIP, and glucagon receptors simultaneously) versus single-target GLP-1 agonists for PCOS. The multi-agonist approach produced superior metabolic improvement across PCOS traits, including insulin sensitivity, androgen levels, and ovarian morphology. The triple agonist outperformed both semaglutide and tirzepatide on composite metabolic endpoints.[9]
Celik et al. (2026) reviewed the evidence linking GLP-1 receptor analogs to metabolic and reproductive function in PCOS. Their analysis documented improvements in insulin sensitivity, weight reduction, androgen levels, menstrual regularity, and ovulation rates across multiple clinical studies. The reproductive improvements were not fully explained by weight loss alone: women who lost the same amount of weight through diet showed smaller improvements in androgen levels and ovulation than those treated with GLP-1 agonists, suggesting a direct hormonal effect beyond caloric balance.[2]
Bo et al. (2025) conducted a comparative efficacy analysis of pharmacological interventions in PCOS. GLP-1 receptor agonists were superior to metformin for improving insulin sensitivity and reducing total testosterone, and comparable to combined oral contraceptives for menstrual regularity but without the thrombotic risk. The network meta-analysis included liraglutide, exenatide, and semaglutide, with semaglutide showing the largest effect sizes for weight reduction and insulin sensitivity improvement.[10] For more on the cardiovascular considerations of GLP-1 treatment, see GLP-1 drugs and heart disease.
The mechanistic distinction between how GLP-1 agonists and metformin affect PCOS helps explain the clinical difference. Metformin reduces hepatic glucose output and modestly improves peripheral insulin sensitivity, indirectly reducing insulin-driven androgen production. GLP-1 agonists additionally slow gastric emptying (reducing postprandial glucose spikes), enhance beta-cell insulin secretion efficiency (reducing the total insulin needed for glucose control), suppress appetite through hypothalamic satiety centers, and now appear to directly modulate GnRH neuron activity. This multi-target pharmacology means GLP-1 agonists intervene at more points in the PCOS peptide network than any single previous therapy. For an overview of how GnRH antagonists work differently in reproductive medicine, see GnRH antagonists in IVF.
Hoteit et al. (2025) reviewed the dual impact of GLP-1 receptor agonists on metabolic and reproductive health in PCOS, synthesizing evidence that GLP-1 agonists improve both domains simultaneously rather than trading one for the other.[1]
Piazza et al. (2025) provided an overview of incretins and polycystic ovaries specifically, documenting how GLP-1 receptor agonists reduce hyperinsulinemia, lower androgen levels, and improve ovulation rates while simultaneously addressing obesity, the most common metabolic comorbidity in PCOS.[11]
Self-Reinforcing Cycles
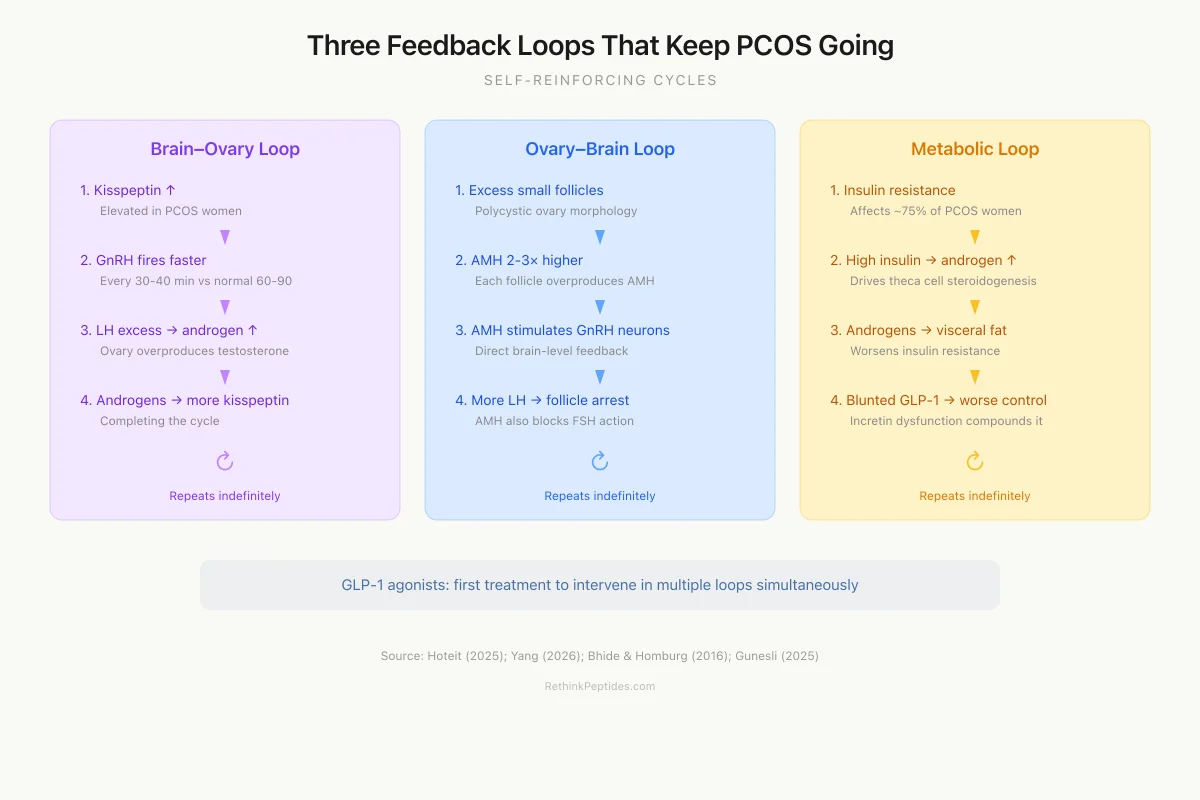
Three Feedback Loops That Keep PCOS Going
Each loop amplifies the others — breaking one can improve them all
Brain–Ovary Loop
Kisspeptin ↑ → GnRH fires faster
GnRH ↑ → LH excess (not enough FSH)
LH ↑ → ovary makes too much testosterone
Testosterone ↑ → stimulates more kisspeptin
Ovary–Brain Loop
Excess small follicles → AMH 2-3× higher
AMH stimulates GnRH neurons directly
GnRH ↑ → more LH → more androgen
AMH also blocks FSH → follicles can't mature
Metabolic–Reproductive Loop
Insulin resistance → high insulin
Insulin drives ovarian androgen production
Androgens → visceral fat → worse insulin resistance
Blunted GLP-1 → worse insulin control
GLP-1 agonists are the first treatment to intervene in multiple loops simultaneously: they reduce insulin (breaking the metabolic loop), normalize GnRH pulsatility (breaking the brain-ovary loop), and improve incretin dynamics.
Source: Hoteit (2025); Yang (2026); Bhide & Homburg (2016)
 View as image
View as imageThe Interconnected Web
What makes PCOS resistant to simple treatments is the interconnection among these peptide pathways. Each dysregulated peptide amplifies the others:
- Elevated kisspeptin accelerates GnRH pulsatility, driving LH excess
- LH excess stimulates ovarian androgen production
- Androgens stimulate kisspeptin expression (completing the loop)
- Excess AMH inhibits FSH-dependent follicle maturation
- AMH also stimulates GnRH neurons directly
- Hyperinsulinemia potentiates ovarian androgen production
- Androgens promote insulin resistance (completing another loop)
- Blunted GLP-1 secretion worsens insulin resistance
- Reduced dynorphin removes the brake on GnRH firing
This network architecture explains why PCOS presents with such heterogeneous phenotypes. The same underlying peptide dysregulation can manifest differently depending on which loops are most active in a given patient. A woman with primarily central (kisspeptin/GnRH) dysfunction may present with anovulation and androgen excess without significant metabolic disease. A woman with primarily metabolic (insulin/incretin) dysfunction may present with obesity and insulin resistance before reproductive symptoms become apparent. Most women have elements of both, but the relative contribution varies.
The network model also explains why PCOS symptoms change across the lifespan. In adolescence, the reproductive axis dysregulation dominates: irregular periods, acne, and hirsutism are the presenting complaints. In the late 20s and 30s, metabolic features become more prominent: weight gain accelerates, insulin resistance worsens, and type 2 diabetes risk increases. By perimenopause, androgen levels naturally decline and menstrual irregularity becomes less distinguishable from normal aging, but the metabolic consequences persist. Cardiovascular disease risk, driven by decades of hyperinsulinemia, dyslipidemia, and chronic low-grade inflammation, becomes the dominant clinical concern. Each phase involves different peptide pathways being most active, but the underlying network dysfunction persists throughout.
Papaetis (2022) reviewed this interconnected pathophysiology in the context of GLP-1 receptor agonist therapy, documenting how interventions at one node in the network propagate effects throughout the system. GLP-1 agonists reduce insulin levels (breaking the insulin-androgen loop), normalize GnRH pulsatility (breaking the kisspeptin-GnRH-LH loop), and improve incretin dynamics (restoring normal meal-related metabolic signaling).[12]
Bader et al. (2024) systematically reviewed GLP-1 effects on anthropometric, metabolic, and endocrine parameters in PCOS, confirming improvements across all three domains and supporting the concept of PCOS as an interconnected peptide hormone disorder rather than separate metabolic and reproductive conditions.[13]
Treatment Approaches
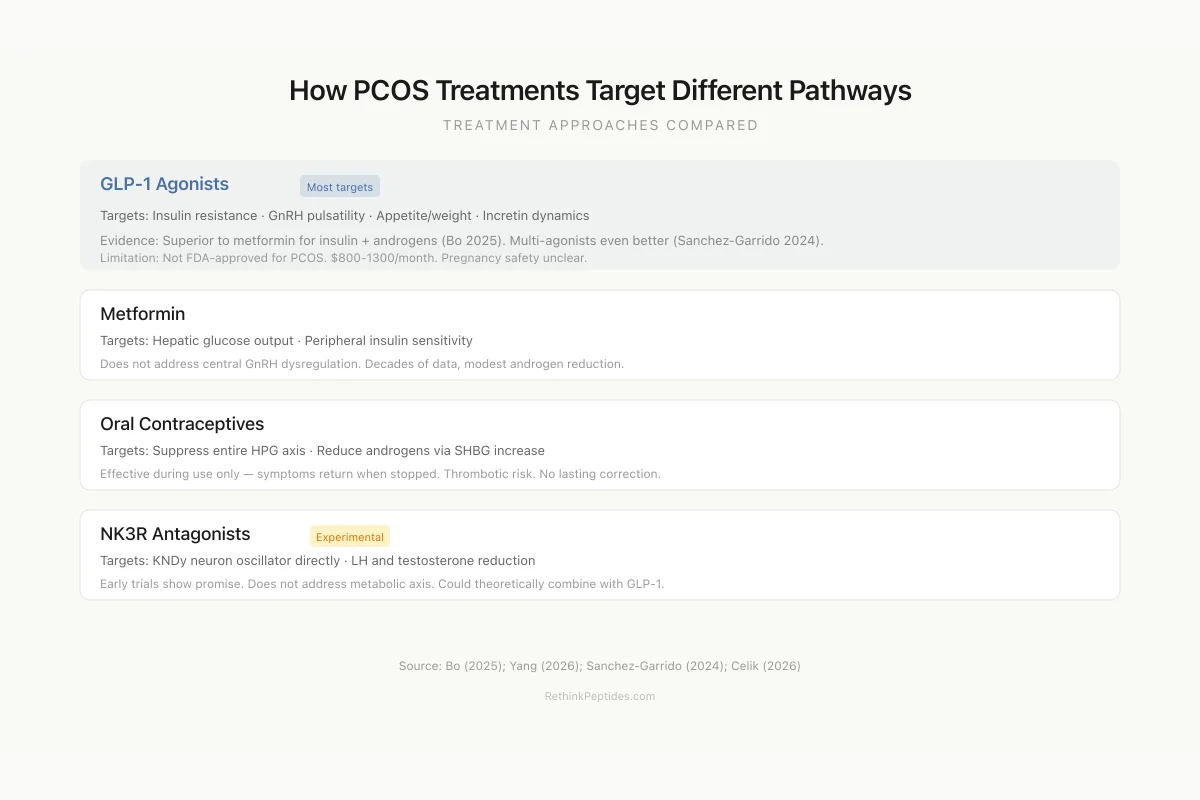
How PCOS Treatments Target Different Peptide Pathways
GLP-1 Agonists Most targets
Evidence: Superior to metformin for insulin + androgens (Bo 2025)
Limitation: Not FDA-approved for PCOS. $800-1300/month.
Metformin
Evidence: Decades of PCOS data, modest androgen reduction
Limitation: Does not address central GnRH dysregulation
Oral Contraceptives
Evidence: Effective for symptoms during use
Limitation: No lasting benefit — symptoms return when stopped. Thrombotic risk.
NK3R Antagonists
Evidence: Early clinical trials show LH/T reduction
Limitation: Experimental. Does not address metabolic axis.
Source: Bo (2025); Yang (2026); Sanchez-Garrido (2024)
 View as image
View as imageCurrent Limitations
The peptide hormone framework for PCOS remains incomplete. Most kisspeptin and KNDy neuron data come from animal models or small human studies; large-scale clinical measurements of hypothalamic peptide activity in PCOS patients are not feasible with current technology. Cerebrospinal fluid sampling and neuroimaging provide indirect evidence but cannot measure GnRH pulse frequency directly in living humans.
The GLP-1 agonist data for PCOS, while promising, come primarily from studies with small sample sizes and short durations. Most published trials enrolled 30-100 participants and followed them for 12-24 weeks. Long-term effects on fertility outcomes (live birth rates, not just ovulation) are poorly characterized. Whether GLP-1 agonists should be continued during conception attempts, and their safety in early pregnancy, remains an active area of investigation. The FDA has not approved any GLP-1 agonist specifically for PCOS; current use is off-label, based on the metabolic overlap between PCOS and type 2 diabetes or obesity indications.
The cost of GLP-1 agonist therapy is a practical barrier to widespread PCOS treatment. Monthly costs for semaglutide or liraglutide range from $800 to $1,300 without insurance coverage, and many insurers restrict coverage to patients with a confirmed diabetes or BMI-based obesity diagnosis. Women with PCOS who are lean or have BMIs below the obesity threshold may not qualify for insurance coverage despite having the metabolic dysregulation that would benefit from treatment. This creates a treatment access disparity based on insurance criteria rather than clinical need. For broader analysis of these economic considerations, see cost-effectiveness of GLP-1s.
The incretin data in PCOS also raise questions about DPP-4 inhibitors (sitagliptin, saxagliptin), which prevent incretin degradation rather than mimicking incretin signaling. If endogenous GLP-1 secretion is blunted in PCOS, DPP-4 inhibitors that preserve what little GLP-1 is produced may be less effective than GLP-1 agonists that provide supraphysiologic stimulation. Limited clinical data support this prediction: DPP-4 inhibitors show modest effects on insulin resistance in PCOS but smaller improvements in androgen levels and ovulation compared to GLP-1 agonists.
Individual variability in PCOS phenotype means that no single peptide intervention will work for all patients. The heterogeneity of the syndrome, with four recognized phenotypes based on combinations of androgen excess, ovulatory dysfunction, and polycystic ovarian morphology, suggests that treatment should be tailored to the dominant peptide dysregulation in each patient. This precision approach requires biomarkers that can identify which peptide loops are most active, a goal that is conceptually clear but practically challenging with current diagnostic tools.
The fertility question remains particularly complex. GLP-1 agonists improve ovulation rates, but they are typically discontinued before conception attempts due to insufficient pregnancy safety data. This creates a treatment gap: the metabolic and hormonal improvements achieved during GLP-1 therapy may partially reverse during the washout period before conception, potentially reducing the fertility benefit. Ongoing clinical trials are investigating whether the reproductive improvements from GLP-1 treatment persist long enough after discontinuation to benefit conception outcomes, but definitive data from large randomized trials with live birth as the primary endpoint are not yet available. For considerations about GLP-1 use in different populations, see GLP-1 weight loss and sarcopenia: the hidden risk in older adults.
The Bottom Line
PCOS is a disorder of interconnected peptide hormone dysregulation spanning the hypothalamus (kisspeptin, GnRH), pituitary (LH/FSH ratio), ovary (AMH, androgens), and metabolic system (insulin, GLP-1, GIP). Each dysregulated peptide amplifies the others through positive feedback loops. GLP-1 receptor agonists have emerged as a treatment that acts on both the central neuroendocrine axis and the metabolic system simultaneously, with multi-agonist approaches showing superior efficacy. The heterogeneity of PCOS phenotypes reflects which peptide loops dominate in individual patients, pointing toward the need for precision treatment strategies.
Sources & References
- 1RPEP-11424·Hoteit, Bassel H et al. (2025). “The dual impact of GLP-1 receptor agonists on metabolic and reproductive health in polycystic ovary syndrome: insights from human and animal trials..” Therapeutic advances in endocrinology and metabolism.Study breakdown →PubMed →↩
- 2RPEP-14947·Celik, Ozlem et al. (2026). “How GLP-1 Drugs Like Semaglutide May Help Women with PCOS and Obesity.” Obesity facts.Study breakdown →PubMed →↩
- 3RPEP-16475·Yang, Linlin et al. (2026). “Hypothalamic RASA1/Ras/AKT/GnRH axis reprogramming mediates GLP-1RA's central rescue of PCOS HPG dysfunction..” Diabetes.Study breakdown →PubMed →↩
- 4RPEP-08375·Hestiantoro, Andon et al. (2024). “Women With PCOS Have Disrupted Kisspeptin-to-Dynorphin Balance.” International journal of reproductive biomedicine.Study breakdown →PubMed →↩
- 5RPEP-02876·Bhide, Priya et al. (2016). “Anti-Müllerian hormone and polycystic ovary syndrome..” Best practice & research. Clinical obstetrics & gynaecology.Study breakdown →PubMed →↩
- 6RPEP-11206·Gunesli, Irmak et al. (2025). “Women with PCOS Have Lower Oxytocin and GLP-1 Levels, Which May Explain Their Stronger Food Cravings.” European journal of endocrinology.Study breakdown →PubMed →↩
- 7RPEP-10171·Bizoń, Anna et al. (2025). “Associations of Serum GIP, GLP-1, and DPP-4 with Metabolic and Hormonal Profiles and Tobacco Exposure in Women with Polycystic Ovary Syndrome..” International journal of molecular sciences.Study breakdown →PubMed →↩
- 8RPEP-06008·Bednarz, Krzysztof et al. (2022). “The Role of Glp-1 Receptor Agonists in Insulin Resistance with Concomitant Obesity Treatment in Polycystic Ovary Syndrome..” International journal of molecular sciences.Study breakdown →PubMed →↩
- 9RPEP-09356·Sánchez-Garrido, Miguel A et al. (2024). “GLP-1/Estrogen Combo Outperforms Other Multi-Agonists for PCOS in Mice.” Nature communications.Study breakdown →PubMed →↩
- 10RPEP-10183·Bo, Yali et al. (2025). “GLP-1 Drugs Added to Standard PCOS Treatment Outperform Standard Therapy Alone for Weight and Metabolic Health.” BMC women's health.Study breakdown →PubMed →↩
- 11RPEP-13048·Piazza, Mauri José (2025). “GLP-1 Medications Show Promise for Women with Polycystic Ovary Syndrome.” JBRA assisted reproduction.Study breakdown →PubMed →↩
- 12RPEP-06418·Papaetis, Georgios S et al. (2022). “GLP-1 receptor agonists, polycystic ovary syndrome and reproductive dysfunction: Current research and future horizons..” Advances in clinical and experimental medicine : official organ Wroclaw Medical University.Study breakdown →PubMed →↩
- 13RPEP-07800·Bader, Salwa et al. (2024). “GLP-1 Drugs for PCOS: What a Systematic Review Says About Weight, Hormones, and Metabolism.” Women's health (London.Study breakdown →PubMed →↩