Kisspeptin: The Peptide That Controls Puberty
Kisspeptin and Reproduction
2003 discovery year
In 2003, two independent research groups discovered that mutations in the kisspeptin receptor GPR54 caused hypogonadotropic hypogonadism, identifying kisspeptin as the missing link between the brain and puberty.
de Roux et al., PNAS, 2003
de Roux et al., PNAS, 2003
If you only read one thing
Kisspeptin is a hormone your brain uses to control your entire reproductive system. Without it, puberty never happens — we know this because people with broken kisspeptin receptors simply don't develop sexually. It's the upstream switch that starts the hormonal cascade leading to testosterone, estrogen, and fertility. Recent clinical trials also show it can boost sexual desire in both men and women. Your body also uses it to decide whether you're eating enough to reproduce — it's the link between nutrition and fertility.
The question of what starts puberty occupied reproductive biologists for decades. The hypothalamic-pituitary-gonadal (HPG) axis was well mapped: gonadotropin-releasing hormone (GnRH) neurons in the hypothalamus release pulses of GnRH, which trigger luteinizing hormone (LH) and follicle-stimulating hormone (FSH) from the pituitary, which drive gonadal development. But what activates the GnRH neurons in the first place? In 2003, de Roux et al. identified the answer: a family in France with hypogonadotropic hypogonadism carried a loss-of-function mutation in GPR54, the receptor for kisspeptin.[1] Without functional kisspeptin signaling, puberty never begins. This discovery redefined kisspeptin from its original identification as a tumor metastasis suppressor (hence its gene name KISS1, from KiSS-1 metastasis-suppressor) into the master upstream regulator of mammalian reproduction. This article examines what two decades of research have revealed about kisspeptin's role in puberty, fertility, sexual behavior, and metabolic regulation. For deeper coverage of kisspeptin's GnRH control mechanism, see Kisspeptin and GnRH: How One Peptide Controls Your Reproductive Hormones. For the sexual behavior evidence, see Kisspeptin and Sexual Arousal: Brain Activation Studies. For therapeutic applications, see Kisspeptin for Hypogonadism: Restoring the Hormonal Cascade. For context on kisspeptin's downstream target, see GnRH: The Master Switch for Reproductive Hormones.
Key Takeaways
- Researchers found families where nobody ever went through puberty — one broken gene was the cause.
- Without kisspeptin, the whole chain of sex hormones never turns on — no puberty, no fertility, nothing.
- In a trial of men with low desire, kisspeptin boosted erection response up to 56% over placebo.
- Kisspeptin also worked in women with low desire — it activates the brain's reproduction circuits directly.
- Most libido drugs tweak blood flow or serotonin. Kisspeptin talks to the brain's actual sexual-desire pathway.
- Your body uses kisspeptin as a "can we afford to reproduce?" check — this is why anorexia stops periods.
- This reproductive switch is so old that even starfish carry a working version of it.
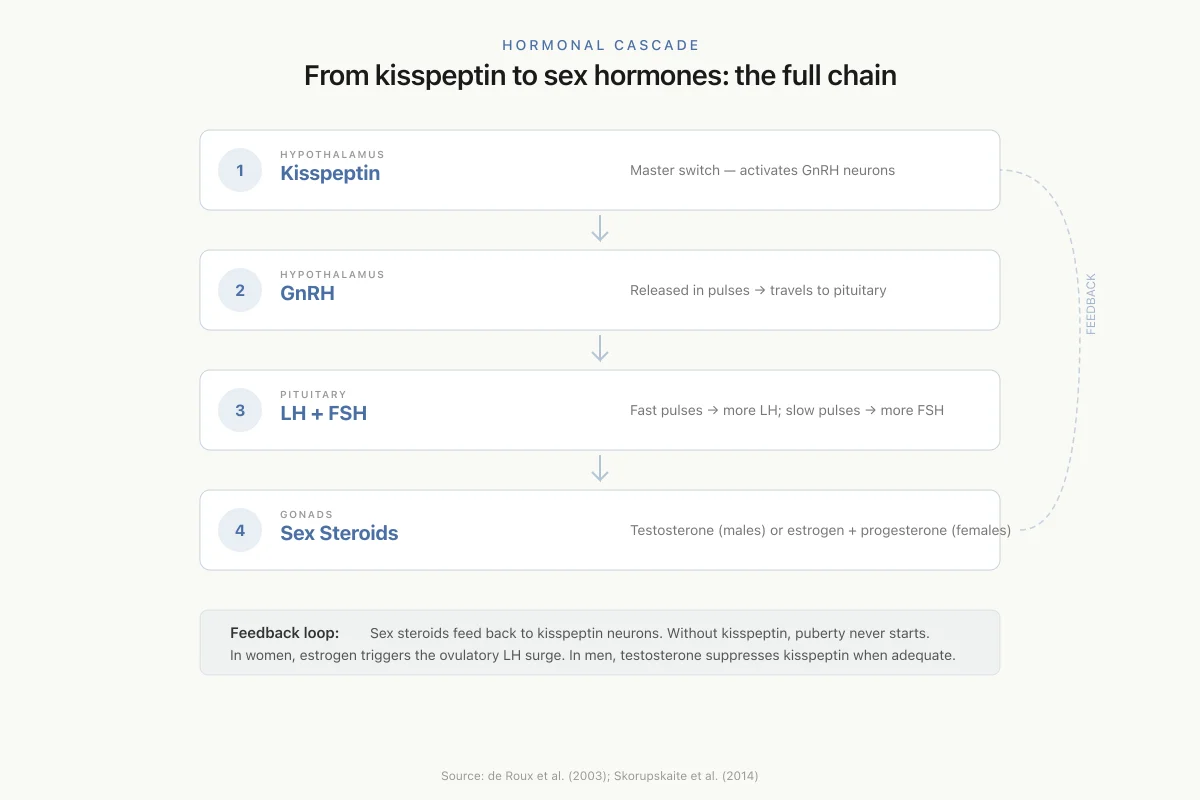
Hormonal Cascade
From kisspeptin to sex hormones: the full chain
Kisspeptin
Master switch — activates GnRH neurons
GnRH
Released in pulses → travels to pituitary
LH + FSH
Fast pulses → more LH; slow pulses → more FSH
Sex Steroids
Testosterone (males) or estrogen + progesterone (females)
Feedback loop: Sex steroids feed back to kisspeptin neurons. In women, estrogen triggers the ovulatory LH surge. In men, testosterone suppresses kisspeptin when levels are adequate. Without kisspeptin, this entire cascade shuts down — puberty never starts.
Source: de Roux et al. (2003); Skorupskaite et al. (2014)
 View as image
View as imageThe Discovery That Changed Reproductive Biology
Before 2003, kisspeptin was known primarily to cancer biologists. The KISS1 gene was identified in 1996 as a metastasis suppressor in melanoma cells, its name a playful reference to Hershey's Kisses chocolates (the gene was discovered at Penn State's Hershey Medical Center). The peptide products of KISS1, called kisspeptins, were later shown to bind GPR54, an orphan G-protein coupled receptor. But neither the cancer biology community nor the reproductive biology community anticipated what came next.
De Roux et al. (2003) studied a consanguineous family in France where multiple members had idiopathic hypogonadotropic hypogonadism: they had normal brain anatomy, no anosmia (ruling out Kallmann syndrome), but complete absence of pubertal development and reproductive function. Genetic analysis revealed a homozygous deletion in GPR54 that abolished receptor function.[1] Independently, Seminara et al. published similar findings in the same year, identifying GPR54 mutations in a Saudi Arabian family with the same phenotype. Two groups, two continents, one conclusion: kisspeptin-GPR54 signaling is non-redundant and indispensable for human puberty and fertility.
Colledge (2008) synthesized the early evidence and clarified the receptor pharmacology. GPR54 (later renamed KISS1R) is expressed on GnRH neurons in the hypothalamus. When kisspeptin binds KISS1R, it triggers a Gq/11-mediated signaling cascade that depolarizes GnRH neurons and stimulates GnRH release. GPR54-knockout mice recapitulate the human phenotype: absent puberty, low gonadotropins, and infertility that is rescued by exogenous GnRH administration, confirming that the defect is upstream of GnRH neurons, not within them.[2]
How Kisspeptin Triggers Puberty
The central question after 2003 was: if kisspeptin activates GnRH neurons, what changes during childhood to initiate the kisspeptin signal? Tena-Sempere (2010) reviewed the emerging evidence for a developmental switch in kisspeptin expression. Kisspeptin neurons in the hypothalamus are present before puberty but express low levels of kisspeptin. At puberty onset, KISS1 gene expression increases dramatically in two hypothalamic populations: the arcuate nucleus (ARC) and the anteroventral periventricular nucleus (AVPV).[3]
The trigger for this increase involves removal of inhibitory restraints rather than addition of new stimulatory signals. During childhood, the hypothalamus is in a state of active suppression: GABAergic, opioidergic, and other inhibitory inputs keep KISS1 expression low. At the onset of puberty, these inhibitory inputs are progressively withdrawn, allowing KISS1 transcription to ramp up. The resulting kisspeptin surge activates GnRH neurons, initiating the pulsatile GnRH secretion that drives pituitary gonadotropin release and gonadal maturation.
The two kisspeptin neuron populations serve distinct functions. The ARC population generates tonic (pulsatile) GnRH secretion that maintains baseline gonadotropin levels throughout life. The AVPV population, which is larger in females, mediates the surge mode of GnRH secretion: the pre-ovulatory LH surge that triggers ovulation. Both populations increase kisspeptin expression at puberty, but the AVPV population's activation is particularly important for the establishment of ovulatory cycles in females.
Roa et al. (2011) summarized the consensus view: kisspeptin acts as a "gatekeeper" of puberty by integrating developmental, metabolic, and environmental signals to determine when reproductive maturation should begin. This gatekeeper function explains clinical observations like delayed puberty in malnourished children and precocious puberty in some conditions of early hypothalamic activation: both involve alterations in kisspeptin signaling timing.[4]
Gain-of-function mutations in KISS1 or KISS1R have been identified in cases of central precocious puberty, providing the mirror image of the loss-of-function hypogonadism described by de Roux. When kisspeptin signaling is constitutively activated, puberty begins too early. This bidirectional genetic evidence, where too little kisspeptin prevents puberty and too much accelerates it, establishes kisspeptin as a true dose-dependent regulator rather than a permissive factor.
The KNDy Neuron: Kisspeptin's Control Circuit
One of the most important developments in kisspeptin biology was the identification of KNDy neurons. Lehman et al. (2010) described a population of neurons in the arcuate nucleus that co-express three neuropeptides: kisspeptin, neurokinin B (NKB), and dynorphin. These KNDy neurons form an autoregulatory circuit that generates the pulsatile pattern of GnRH secretion essential for normal reproductive function.[5]
The model works as follows: NKB acts on other KNDy neurons through NK3 receptors to stimulate kisspeptin release (the "on" signal). Kisspeptin then activates nearby GnRH neurons through KISS1R. Simultaneously, NKB stimulates dynorphin release, which acts through kappa-opioid receptors to inhibit kisspeptin output (the "off" signal). This NKB-stimulation/dynorphin-inhibition cycle produces the rhythmic pulses of kisspeptin that drive pulsatile GnRH secretion.
Navarro (2015) expanded the KNDy model to include tachykinin interactions beyond NKB. The integrated hypothalamic tachykinin-kisspeptin system involves substance P and other tachykinins that modulate the KNDy circuit, adding further regulatory layers. These tachykinin inputs allow the GnRH pulse generator to respond to a broader range of physiological signals, including stress, inflammation, and circadian rhythms.[6]
Pulse Generator
How KNDy neurons create the GnRH pulse
NKB acts on NK3 receptors on neighboring KNDy neurons, stimulating kisspeptin release
Kisspeptin binds KISS1R on GnRH neurons → GnRH pulse → LH/FSH release from pituitary
Simultaneously, NKB triggers dynorphin release. Dynorphin acts on kappa-opioid receptors to suppress kisspeptin output
With kisspeptin suppressed, GnRH pauses. As dynorphin effect wears off, NKB fires again → next pulse
Source: Lehman et al. (2010), Endocrinology
 View as image
View as imageRackova et al. (2025) reviewed the current understanding of how each component of the KNDy system contributes to reproductive regulation. The balance between NKB (stimulatory) and dynorphin (inhibitory) inputs determines pulse frequency, and this frequency encodes the signal that the pituitary reads: fast pulses favor LH secretion, slow pulses favor FSH. Disruptions to this balance underlie reproductive disorders including hypothalamic amenorrhea (too little pulsatility) and polycystic ovary syndrome (too much).[7]
Kisspeptin and Sexual Desire
Beyond its reproductive endocrine functions, kisspeptin has emerged as a modulator of sexual behavior and desire. The Dhillo laboratory at Imperial College London conducted two landmark randomized controlled trials investigating kisspeptin's effects on sexual brain processing.
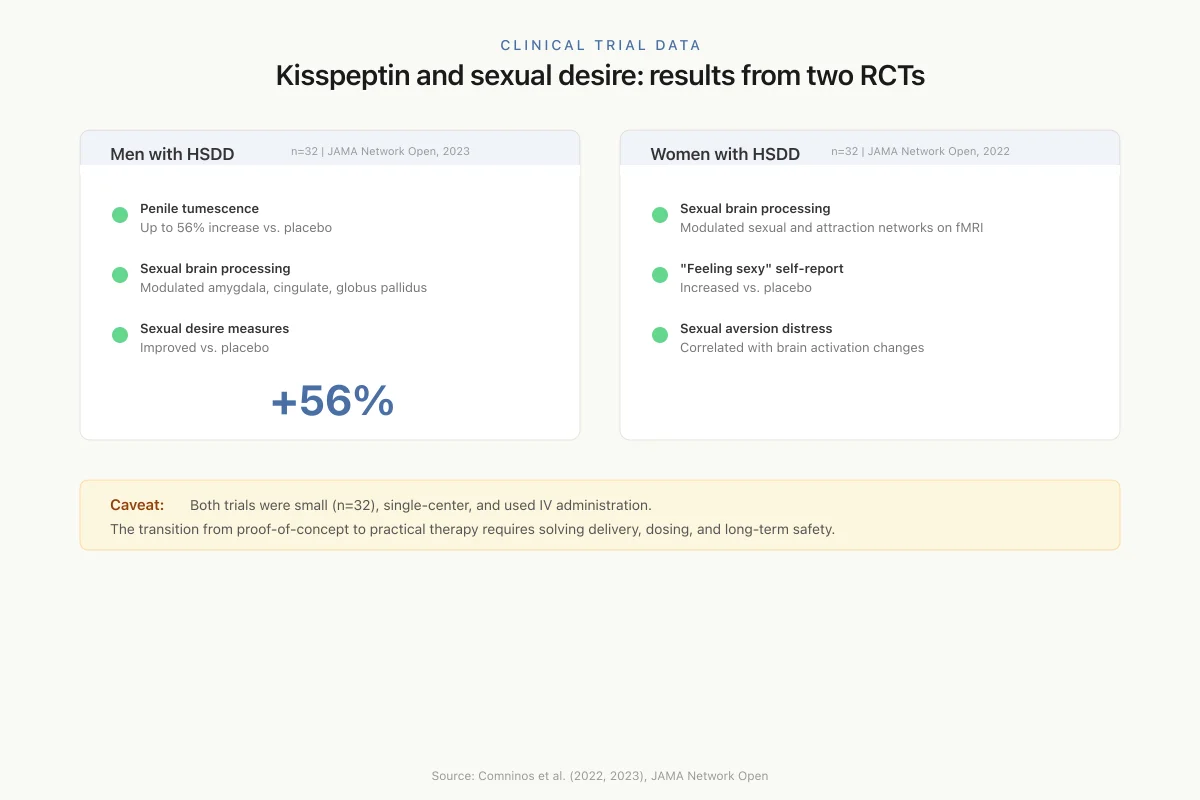
In a double-blind crossover trial published in JAMA Network Open (2023), 32 men with hypoactive sexual desire disorder (HSDD) received intravenous kisspeptin infusion (1 nmol/kg/h for 75 minutes) or placebo. Kisspeptin modulated brain activity in key structures of the sexual-processing network, including the amygdala, cingulate cortex, and globus pallidus. Penile tumescence in response to sexual stimuli increased by up to 56% above placebo. Behavioral measures of sexual desire and arousal also improved.[8]
A parallel trial in 32 premenopausal women with HSDD, published in JAMA Network Open (2022), found that kisspeptin modulated sexual and attraction brain processing in functional neuroimaging. Post hoc analysis showed increased self-reported ratings of "feeling sexy" compared with placebo. The effects correlated with psychometric measures of sexual aversion and associated distress.[9]
Clinical Trial Data
Kisspeptin and sexual desire: results from two RCTs
Men with HSDD
n=32 | JAMA Network Open, 2023Women with HSDD
n=32 | JAMA Network Open, 2022Caveat: Both trials were small (n=32), single-center, and used IV administration. The transition from proof-of-concept to practical therapy requires solving delivery, dosing, and long-term safety.
Source: Comninos et al. (2023); Comninos et al. (2022), JAMA Network Open
 View as image
View as imageBoth trials reported that kisspeptin administration was well-tolerated with no adverse effects. Mills and Dhillo (2022) placed these findings in context: kisspeptin may represent a fundamentally new pharmacological approach to sexual desire disorders because it engages the brain's reproductive-behavioral circuits rather than simply modulating peripheral blood flow or neurotransmitter levels, as existing treatments attempt to do.[10]
These results are preliminary. The sample sizes were small (32 participants per trial), the kisspeptin was administered intravenously (impractical for routine use), and the effects were measured acutely rather than over chronic administration. The transition from proof-of-concept to clinical therapy requires solving delivery, dosing, and long-term safety questions. However, the consistent modulation of sexual brain processing across both sexes supports a genuine central effect rather than a peripheral artifact.
The Metabolism-Reproduction Connection
Kisspeptin neurons do not operate in isolation from metabolic signals. Navarro (2011) documented that kisspeptin neurons in the arcuate nucleus express receptors for leptin (the adiposity signal) and insulin, positioning them to integrate nutritional status with reproductive output.[11]
This integration has direct clinical relevance. In states of severe energy deficit (anorexia nervosa, excessive exercise, famine), leptin levels fall, kisspeptin expression decreases, GnRH pulsatility slows, and reproductive function ceases. The body effectively shuts down reproduction when energy is insufficient to sustain pregnancy and lactation. Conversely, in obesity, elevated leptin was initially expected to boost kisspeptin, but the relationship is more nuanced: leptin resistance in obese individuals may impair kisspeptin signaling despite high circulating leptin levels.
Sliwowska et al. (2024) extended this metabolic connection to diabetes and metabolic syndrome. Their review documented evidence that kisspeptin acts not only as a reproductive neuropeptide but also as a direct metabolic regulator, influencing insulin secretion, glucose homeostasis, and adipose tissue function. In pancreatic beta cells, kisspeptin modulates insulin release through KISS1R signaling, providing a direct endocrine link between the reproductive and metabolic systems that does not require hypothalamic mediation. The effects are sexually dimorphic: kisspeptin's metabolic actions differ between males and females, complicating therapeutic development. In animal models, kisspeptin administration improved glucose tolerance in male but not female mice, while reproductive responses showed the opposite pattern.[12]
This bidirectional relationship between metabolism and kisspeptin has clinical implications beyond fertility. Type 2 diabetes, metabolic syndrome, and obesity are all associated with reproductive dysfunction (hypogonadism in men, menstrual irregularity in women), and kisspeptin dysregulation may be a common mechanistic link. The observation that weight loss can restore menstrual regularity in obese women with anovulation may be mediated in part through restored kisspeptin signaling as leptin sensitivity improves.
Skorupskaite et al. (2014) mapped the full kisspeptin-GnRH pathway in human health and disease, documenting how sex steroid feedback (both estrogen and testosterone) acts through kisspeptin neurons to regulate GnRH secretion. In women, estrogen's positive feedback on the AVPV kisspeptin population triggers the pre-ovulatory LH surge that causes ovulation. In men, testosterone's negative feedback on ARC kisspeptin neurons maintains the gonadal thermostat: when testosterone falls, kisspeptin increases, driving more GnRH and LH to restore testosterone production. Disruption of this feedback loop at the kisspeptin level can explain anovulation in conditions ranging from hypothalamic amenorrhea to PCOS, and may contribute to hypogonadism in aging men.[13]
The sex steroid feedback system also explains why kisspeptin is central to understanding the menstrual cycle. The mid-cycle estrogen rise from the dominant ovarian follicle triggers the AVPV kisspeptin surge, which drives the GnRH/LH surge causing ovulation. After ovulation, progesterone from the corpus luteum suppresses kisspeptin, preventing a second LH surge. This monthly cycle of kisspeptin activation and suppression represents one of the most dynamic neuropeptide fluctuations in the human body.
Kisspeptin in Polycystic Ovary Syndrome
Polycystic ovary syndrome (PCOS) affects 8-13% of reproductive-age women and is characterized by hyperandrogenism, anovulation, and increased LH pulse frequency. Hestiantoro et al. (2024) conducted a cross-sectional study comparing kisspeptin and dynorphin expression in PCOS patients versus controls. PCOS patients showed altered neuropeptide expression patterns, with elevated kisspeptin and reduced dynorphin levels that would be expected to increase GnRH/LH pulsatility.[14]
The KNDy model provides a framework for understanding this imbalance. If kisspeptin input is elevated (more "on" signal) and dynorphin input is reduced (less "off" signal), the net effect is faster GnRH pulsatility, more LH relative to FSH, and the androgenic, anovulatory state characteristic of PCOS. Therapeutic strategies targeting this imbalance, including NK3 receptor antagonists that reduce NKB-driven kisspeptin release, are in clinical development. Mills and Dhillo (2022) reviewed these emerging therapies, noting that NK3 receptor antagonists have shown promise in early-phase trials for reducing LH pulse frequency and testosterone levels in PCOS patients.[10]
An Evolutionarily Ancient System
Kisspeptin signaling is not a recent vertebrate innovation. Islam et al. (2026) demonstrated functional kisspeptin-type peptides in echinoderms (sea urchins and starfish), organisms that diverged from the vertebrate lineage over 500 million years ago. The echinoderm kisspeptin-type peptides activated reproductive behaviors and gonadal functions analogous to those in vertebrates, suggesting deep evolutionary conservation of this signaling system.[15]
This evolutionary depth is relevant for understanding kisspeptin's fundamental biology. A system conserved for half a billion years is not a peripheral add-on to reproduction: it is architecturally central. The conservation also suggests that kisspeptin's roles may extend beyond what has been characterized in mammalian models, as the signaling system predates the emergence of the mammalian HPG axis in its current form.
The evolutionary perspective also illuminates kisspeptin's multi-functionality. In echinoderms, kisspeptin-type peptides influence not only gonadal function but also feeding behavior and energy allocation, functions that parallel the metabolic-reproductive integration seen in mammalian kisspeptin neurons. This suggests that kisspeptin's role as a metabolic-reproductive integrator is not a secondary adaptation but an ancestral function, predating the evolution of the HPG axis itself.
Limitations of Current Research
The human evidence for kisspeptin's role in puberty comes primarily from loss-of-function genetics. While the GPR54 mutation families definitively establish kisspeptin signaling as necessary for puberty, they do not fully characterize the normal physiological dynamics of kisspeptin during pubertal maturation. Longitudinal studies measuring kisspeptin levels across puberty in healthy children are limited by ethical and practical constraints.
The sexual desire trial data, while from randomized controlled trials, involves small sample sizes, acute intravenous administration, and single-center designs. The HSDD diagnostic category itself is debated within psychiatry and sexual medicine. Whether kisspeptin's acute effects on brain imaging and penile tumescence translate to clinically meaningful improvements in sexual satisfaction during chronic outpatient use remains unknown.
The PCOS data is cross-sectional and cannot establish whether altered kisspeptin/dynorphin expression is a cause or consequence of the PCOS hormonal milieu. Animal models support a causative role for kisspeptin dysregulation in PCOS-like phenotypes, but the human genetics of PCOS are complex and polygenic.
Most mechanistic work on kisspeptin signaling, including the KNDy model, derives from rodent studies. While the core pathway is conserved in humans, there are species differences in kisspeptin neuron distribution, receptor expression patterns, and the relative contributions of different hypothalamic nuclei. For example, the AVPV kisspeptin population that mediates the LH surge in rodents has a less clearly defined anatomical equivalent in humans, where the surge mechanism may involve the preoptic area. Translating rodent KNDy circuit dynamics to human neuroendocrine physiology requires caution, and human-specific studies remain essential for validating therapeutic targets derived from animal models.
Therapeutic Landscape
The therapeutic potential of kisspeptin targets three areas: reproductive disorders, sexual health, and metabolic disease. Mills and Dhillo (2022) reviewed the translational pipeline, noting that the kisspeptin/NKB/dynorphin system offers multiple pharmacological entry points. Direct kisspeptin agonists could potentially treat conditions of insufficient GnRH pulsatility (hypothalamic amenorrhea, male hypogonadism). NK3 receptor antagonists could reduce excessive GnRH pulsatility in PCOS and endometriosis. And kisspeptin itself, or analogs with improved pharmacokinetics, could address sexual desire disorders through central brain mechanisms.[10]
The practical challenges are substantial. Native kisspeptin has a short half-life (minutes) and requires intravenous administration. Developing kisspeptin analogs with oral bioavailability or subcutaneous depot formulations is an active area of pharmaceutical research. Several approaches are under investigation: modified kisspeptin peptides with enhanced protease resistance, small-molecule KISS1R agonists that could be taken orally, and NK3 receptor antagonists that modulate kisspeptin output indirectly.
The sexually dimorphic effects of kisspeptin on metabolism and reproduction mean that therapies may need to be sex-specific. And the tight integration of kisspeptin with the entire HPG axis means that manipulating kisspeptin signaling will inevitably affect multiple downstream hormone systems. A kisspeptin agonist intended to treat female sexual desire would also stimulate GnRH, LH, FSH, and sex steroid production, effects that would need to be carefully managed. This distinguishes kisspeptin from peripheral sexual health treatments that have fewer systemic endocrine consequences.
The NK3 receptor antagonist approach sidesteps some of these challenges. By modulating NKB signaling within the KNDy circuit rather than directly activating KISS1R, these drugs can fine-tune GnRH pulsatility without maximally stimulating the pathway. Early clinical data from several pharmaceutical companies has shown that NK3 antagonists can reduce LH pulse frequency and lower testosterone in PCOS, reduce hot flushes in menopause, and potentially offer a new mechanism for endometriosis management. For more on how GnRH-based therapies are already used in fertility, see GnRH Antagonists in IVF. For the broader landscape of peptide biomarkers in fertility, see AMH: The Peptide Biomarker for Fertility. For how GnRH agonists treat estrogen-driven conditions, see GnRH Agonists for Endometriosis.
The Bottom Line
Kisspeptin is the upstream master regulator of mammalian reproduction, essential for puberty onset, pulsatile GnRH secretion, and fertility. The KNDy neuron system (kisspeptin/neurokinin B/dynorphin) in the arcuate nucleus generates the GnRH pulse pattern that controls gonadotropin release. Clinical trial data supports kisspeptin's role in modulating sexual desire in both men and women. Metabolic integration through leptin and insulin receptors on kisspeptin neurons links nutritional status to reproductive capacity. Therapeutic targeting of the kisspeptin system is under active clinical development for PCOS, sexual desire disorders, and fertility treatment.
Sources & References
- 1RPEP-00808·de Roux, Nicolas et al. (2003). “Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54..” Proceedings of the National Academy of Sciences of the United States of America.Study breakdown →PubMed →↩
- 2RPEP-01325·Colledge, W H (2008). “Kisspeptin: The Brain's Master Switch for Puberty and Fertility Through GPR54.” Results and problems in cell differentiation.Study breakdown →PubMed →↩
- 3RPEP-01702·Tena-Sempere, Manuel (2010). “Kisspeptin Brain Signaling: The Master Controller of Puberty and Fertility Updated.” Molecular and cellular endocrinology.Study breakdown →PubMed →↩
- 4RPEP-01842·Roa, Juan et al. (2011). “Kisspeptins in Reproductive Biology: 2011 Consensus and Latest Developments.” Biology of reproduction.Study breakdown →PubMed →↩
- 5RPEP-01646·Lehman, Michael N et al. (2010). “Minireview: kisspeptin/neurokinin B/dynorphin (KNDy) cells of the arcuate nucleus: a central node in the control of gonadotropin-releasing hormone secretion..” Endocrinology.Study breakdown →PubMed →↩
- 6RPEP-02753·Navarro, Víctor M et al. (2015). “All Three Tachykinin Peptides — Not Just NKB — Regulate Reproductive Hormones Through Kisspeptin Neurons.” Endocrinology.Study breakdown →PubMed →↩
- 7RPEP-13160·Racková, Jana (2025). “How KNDy Neurons Control Reproduction Through Kisspeptin, Neurokinin B, and Dynorphin.” Ceska gynekologie.Study breakdown →PubMed →↩
- 10RPEP-06371·Mills, Edouard G et al. (2022). “Kisspeptin and Neurokinin B: From Reproductive Brain Science to New Fertility and Menopause Treatments.” Journal of neuroendocrinology.Study breakdown →PubMed →↩
- 11RPEP-01825·Navarro, Victor M et al. (2011). “Kisspeptin Links Metabolism to Fertility: How Nutritional Status Controls Reproduction.” Nature reviews. Endocrinology.Study breakdown →PubMed →↩
- 12RPEP-09287·Sliwowska, Joanna Helena et al. (2024). “Kisspeptin: A Single Peptide That Links Fertility and Metabolism, with Therapeutic Potential for Both.” Journal of diabetes.Study breakdown →PubMed →↩
- 13RPEP-02505·Skorupskaite, Karolina et al. (2014). “How Kisspeptin Controls Your Reproductive Hormones — And Why It Matters for Fertility.” Human reproduction update.Study breakdown →PubMed →↩
- 14RPEP-08375·Hestiantoro, Andon et al. (2024). “Women With PCOS Have Disrupted Kisspeptin-to-Dynorphin Balance.” International journal of reproductive biomedicine.Study breakdown →PubMed →↩
- 15RPEP-15357·Islam, Tabinda et al. (2026). “Kisspeptin-Type Peptides: Ancient Neurohormones Now Understood to Control Reproduction Across Species.” BMC biology.Study breakdown →PubMed →↩