Amyloid-Beta: The Alzheimer's Peptide Explained
Neurodegenerative Peptides
27% slower decline
Lecanemab slowed cognitive decline by 27% versus placebo over 18 months in the CLARITY AD trial of 1,795 people with early Alzheimer's disease.
van Dyck et al., NEJM, 2023
van Dyck et al., NEJM, 2023
If you only read one thing
Amyloid-beta is a tiny protein fragment that builds up in the brains of people with Alzheimer's disease. For 30 years, scientists believed it was the main cause — but over 30 drugs targeting it failed in clinical trials. Then in 2023-2024, two drugs finally showed they could slow the disease by clearing amyloid, earning FDA approval. The catch: they only slow decline by about 27-35%, they cause brain swelling in up to 1 in 4 patients, and they cost over $26,000 a year. It's a breakthrough, but a modest one.
No peptide in science has generated more controversy than amyloid-beta. For over three decades, the "amyloid cascade hypothesis" has dominated Alzheimer's disease research, directing billions of dollars toward the idea that this small peptide fragment, just 36-43 amino acids long, is the central driver of the most common form of dementia. Dozens of clinical trials targeting amyloid-beta failed before lecanemab became the first anti-amyloid antibody to show clear clinical benefit in 2023, slowing cognitive decline by 27% over 18 months.[1] This article covers what amyloid-beta is, how it forms, why it may cause harm, the drugs that target it, and the ongoing debate about whether removing it is enough to stop Alzheimer's. For related topics, see our articles on peptide approaches to Alzheimer's disease, peptide vaccines for Alzheimer's, alpha-synuclein in Parkinson's, NAP peptide (davunetide), and Huntington's disease peptide research.
Key Takeaways
- Amyloid-beta is a tiny protein fragment that builds up in the brains of people with Alzheimer's disease.
- Everyone's brain makes it every day. The problem isn't making it — it's when the cleanup crew can't keep up and sticky clumps start damaging brain cells.
- For 30 years, the amyloid theory drove most Alzheimer's research. Over 30 drugs targeting it failed in trials before any worked.
- In 2023 and 2024, two drugs finally showed they could slow decline by clearing amyloid — and earned FDA approval.
- The catch: they slow progression by only about 27% to 35%. They don't reverse it, and they don't cure the disease.
- Side effects are serious. Brain swelling occurs in up to 1 in 4 patients. The drugs cost over $26,000 a year.
- A surprise: amyloid-beta may actually have a normal job as a brain antimicrobial. Clearing it completely might not be as clean as once hoped.
What Is Amyloid-Beta?
Amyloid-beta (often abbreviated as Abeta or A-beta) is a peptide fragment derived from a larger protein called amyloid precursor protein (APP). APP is a transmembrane protein expressed throughout the body, with the highest levels in the brain. Its normal function is not fully understood, but it appears to play roles in cell adhesion, synapse formation, and neural development.[2]
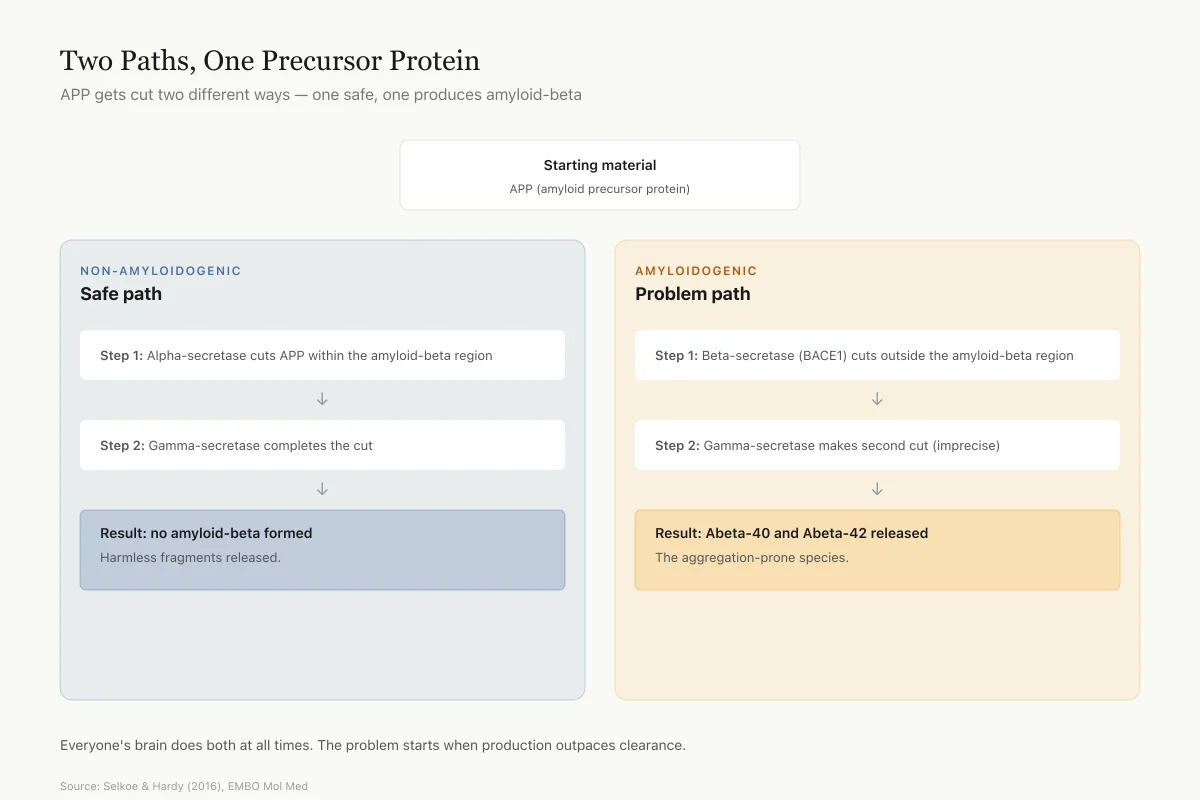
APP can be processed through two different pathways. In the non-amyloidogenic pathway, an enzyme called alpha-secretase cuts APP within the amyloid-beta sequence, preventing amyloid-beta formation. In the amyloidogenic pathway, two different enzymes act sequentially: beta-secretase (BACE1) makes the first cut, and gamma-secretase makes the second, releasing the amyloid-beta peptide from the membrane.[2]
The gamma-secretase cut is imprecise, producing fragments of different lengths. The two most common are Abeta-40 (40 amino acids) and Abeta-42 (42 amino acids). Those extra two amino acids on Abeta-42 make a disproportionate difference: Abeta-42 is far more prone to aggregation and is the dominant species found in amyloid plaques.[2]
APP Processing
Two Paths, One Precursor Protein
The same protein (APP) gets cut two different ways — one safe, one produces amyloid-beta
Starting material
APP (amyloid precursor protein) — transmembrane protein in brain cells
Non-amyloidogenic
Safe path
Amyloidogenic
Problem path
Everyone's brain does both at all times. The problem starts when the amyloidogenic path outpaces clearance. The 2-amino-acid difference between Abeta-40 and Abeta-42 is huge — Abeta-42 clumps far more readily and dominates the plaques.
Source: Selkoe & Hardy (2016), EMBO Mol Med — amyloid cascade hypothesis review
 View as image
View as imageAmyloid-beta is not inherently pathological. Healthy brains produce it continuously, and it is cleared through multiple pathways including enzymatic degradation, transport across the blood-brain barrier, and drainage through the brain's lymphatic-like glymphatic system. Disease occurs when the balance between production and clearance tips, and amyloid-beta accumulates beyond the brain's capacity to remove it.
The Amyloid Cascade Hypothesis
In 1992, John Hardy and Gerald Higgins proposed the amyloid cascade hypothesis, arguing that amyloid-beta accumulation is the initial pathological event in Alzheimer's disease, triggering a downstream cascade of tau hyperphosphorylation, neurofibrillary tangles, neuroinflammation, synaptic loss, and neuronal death.
Selkoe and Hardy (2016) revisited the hypothesis at its 25th anniversary. They argued that genetic, biomarker, and pathological evidence still supported amyloid-beta as the initiating factor, while acknowledging that the hypothesis had been refined over time. Key supporting evidence includes:[2]
- All known genetic causes of early-onset Alzheimer's (mutations in APP, PSEN1, PSEN2) increase amyloid-beta production or the ratio of Abeta-42 to Abeta-40
- Down syndrome (trisomy 21) causes Alzheimer's pathology because the APP gene is on chromosome 21, leading to 50% more APP production
- A protective APP mutation (A673T, the "Icelandic mutation") reduces amyloid-beta production and lowers Alzheimer's risk by 5-7 fold
- Amyloid plaques appear 15-20 years before symptoms, consistent with amyloid as an early trigger
The hypothesis has evolved from "amyloid plaques cause Alzheimer's" to a more nuanced version: soluble amyloid-beta oligomers (small clusters of a few peptides, not the large plaques visible on brain imaging) may be the primary toxic species. Plaques may actually be a relatively inert storage form, while the oligomers circulating between cells cause the real synaptic damage.[2]
Genetics and Risk Factors
The genetics of amyloid-beta are among the strongest pieces of evidence supporting its role in Alzheimer's. Three genes cause autosomal dominant early-onset Alzheimer's: APP (the amyloid precursor protein itself), PSEN1, and PSEN2 (which encode components of gamma-secretase). All three increase either total amyloid-beta production or the ratio of the more aggregation-prone Abeta-42. People with these mutations develop Alzheimer's with near-100% certainty, usually between ages 30 and 60.[2]
For late-onset Alzheimer's (the common form, after age 65), the strongest genetic risk factor is the APOE4 allele. Carrying one copy of APOE4 increases Alzheimer's risk roughly 3-fold; carrying two copies increases it 8-12 fold. APOE4 impairs amyloid-beta clearance from the brain, leading to greater accumulation. It also affects lipid metabolism, neuroinflammation, and blood-brain barrier integrity, suggesting its risk extends beyond amyloid alone.[2]
APOE4 status has become clinically relevant because it affects safety in anti-amyloid therapy. In both the lecanemab and donanemab trials, APOE4 carriers had substantially higher rates of ARIA (brain swelling and microbleeds). APOE4 homozygotes (two copies) had the highest risk of all.
Detecting Amyloid-Beta: Biomarkers
Brain imaging
Amyloid PET scans use radioactive tracers that bind to amyloid plaques, producing images showing plaque distribution and density. This technology revealed that amyloid accumulation begins 15-20 years before cognitive symptoms, during a stage now called "preclinical Alzheimer's." PET scans are the gold standard for confirming amyloid status and are required for enrollment in anti-amyloid clinical trials.
Blood tests
A major recent advance is the development of blood-based amyloid biomarkers. Plasma Abeta-42/Abeta-40 ratio tests can detect brain amyloid pathology with approximately 85-90% accuracy compared to PET scans. These blood tests cost a fraction of PET imaging and can be used for screening in primary care, though they are not yet widely available outside research settings.
Plasma phosphorylated tau (p-tau217 and p-tau181) tests detect both amyloid and tau pathology and may become the preferred first-line screening tools. Their accuracy approaches that of PET scans for identifying people who would benefit from anti-amyloid therapy.
How Amyloid-Beta Damages the Brain
Oligomer toxicity
The shift from plaques to oligomers as the key toxic species has been one of the most important revisions to amyloid biology. Soluble amyloid-beta oligomers bind to synapses, disrupting the signaling between neurons. They impair long-term potentiation (the cellular mechanism underlying learning and memory) and promote long-term depression (weakening of synaptic connections).
Oligomers also trigger neuroinflammation by activating microglia, the brain's immune cells. Chronically activated microglia release inflammatory cytokines that damage surrounding neurons and contribute to the progressive nature of the disease. This inflammatory cascade may be self-sustaining even if amyloid-beta is later removed, which could partly explain why late-stage amyloid removal has limited clinical benefit.
The tau connection
Amyloid-beta accumulation appears to trigger tau pathology. Tau is a protein that normally stabilizes microtubules inside neurons. In Alzheimer's disease, tau becomes hyperphosphorylated, detaches from microtubules, and forms neurofibrillary tangles inside neurons. The density and distribution of tau tangles correlate more closely with cognitive decline than amyloid plaque burden does.[2]
The donanemab TRAILBLAZER-ALZ 2 trial confirmed this relationship: patients with low-to-medium tau pathology (earlier disease stage) benefited more from amyloid removal than patients with high tau pathology (more advanced disease).[4] This suggests that once tau pathology is extensive, removing amyloid may be too late to substantially change the disease course.
Anti-Amyloid Drug Trials: The Full History
Three decades of failure
Jeremic et al. (2021) published a systematic review cataloguing therapeutic strategies against amyloid-beta. The history is sobering: over 30 clinical trials targeting amyloid-beta through various mechanisms (gamma-secretase inhibitors, beta-secretase inhibitors, active vaccines, passive immunotherapy, anti-aggregation compounds) failed to show clinical benefit.[5]
The failures included semagacestat (a gamma-secretase inhibitor that worsened cognition), bapineuzumab and solanezumab (anti-amyloid antibodies that cleared plaques but did not slow decline), and AN1792 (an active vaccine that caused brain inflammation in 6% of participants). Each failure raised the question: is amyloid the wrong target, or were the drugs too weak, given too late, or targeting the wrong form of amyloid?
Lecanemab: CLARITY AD
The landscape changed with lecanemab (brand name Leqembi). Van Dyck and colleagues (2023) published the CLARITY AD trial in the New England Journal of Medicine. This phase 3 trial randomized 1,795 participants with early Alzheimer's disease (mild cognitive impairment or mild dementia with confirmed amyloid pathology) to lecanemab or placebo for 18 months.[1]
Lecanemab targets soluble amyloid-beta protofibrils (larger aggregates than oligomers but smaller than plaques), a form hypothesized to be particularly toxic. The results:
- Primary endpoint: CDR-SB (Clinical Dementia Rating-Sum of Boxes) changed by 1.21 points with lecanemab versus 1.66 with placebo, a difference of -0.45 points (27% less decline, p<0.001)
- Amyloid clearance: Lecanemab reduced amyloid PET signal by an average of 55.48 centiloids, effectively clearing plaques in most patients
- Secondary endpoints: All favored lecanemab, including ADAS-cog14 (-1.44, p<0.001) and activities of daily living measures
The safety profile was the primary concern. Amyloid-related imaging abnormalities (ARIA) occurred in 21.3% of lecanemab patients: ARIA-E (brain edema/effusions) in 12.6% and ARIA-H (brain microbleeds) in 17.3%. Most ARIA events were asymptomatic, detected only on MRI monitoring. Three deaths in the lecanemab group were possibly related to treatment, including two cerebral hemorrhages in patients taking anticoagulants.[1]
The FDA granted traditional approval to lecanemab in July 2023.
Safety
ModerateARIA — amyloid-related imaging abnormalities
Concern
Both lecanemab and donanemab cause ARIA in 12–24% of patients: ARIA-E (brain edema/swelling) and ARIA-H (microbleeds). Most cases are asymptomatic and detected only on MRI, but symptomatic cases occur and deaths have been reported — particularly in patients on anticoagulants or carrying two APOE4 alleles.
What the research says
ARIA usually resolves without long-term consequences when caught on MRI and managed with dose adjustment or pause. The APOE4 homozygote risk is high enough that some clinicians test before starting treatment and weigh that result against expected benefit.
Particularly relevant for: Anyone considering anti-amyloid therapy, especially APOE4 carriers and patients on blood thinners
What to do
Genetic testing for APOE4 status before starting. Regular MRI monitoring during treatment. Review and potentially discontinue anticoagulants with the prescribing physician.
van Dyck et al. (2023) CLARITY AD; Sims et al. (2023) TRAILBLAZER-ALZ 2 safety data
Donanemab: TRAILBLAZER-ALZ 2
Sims and colleagues (2023) published the TRAILBLAZER-ALZ 2 trial in JAMA. This trial randomized 1,736 participants with early symptomatic Alzheimer's disease to donanemab or placebo for 76 weeks. Donanemab targets a modified form of amyloid-beta (pyroglutamate Abeta) found specifically in plaques.[4]
Key results in the low/medium tau population:
- Primary endpoint: 35% slowing of cognitive decline on the iADRS (integrated Alzheimer's Disease Rating Scale)
- No progression: 47% of donanemab patients showed no clinical progression at 1 year, versus 29% on placebo
- Amyloid clearance: 80% of donanemab patients achieved amyloid clearance (less than 24.1 centiloids) by 76 weeks, at which point treatment was stopped
In the combined population (including high-tau patients), the benefit was smaller: 22% slowing of decline. ARIA-E occurred in 24.0% of donanemab patients, with 6.1% being symptomatic. Three deaths were considered treatment-related.[4]
The FDA approved donanemab (brand name Kisunla) in July 2024.
The Debate: Is Amyloid Enough?
The clinical benefits of lecanemab and donanemab validated that amyloid-beta is involved in Alzheimer's progression. But the magnitude of benefit has fueled debate.
A 27% reduction in the rate of decline (lecanemab) translates to roughly 5 months of delayed progression over 18 months. Patients still decline; they decline somewhat more slowly. Whether this degree of benefit is clinically meaningful to patients and families is a genuine question, not a rhetorical one. Some patients and clinicians view any slowing of an otherwise relentless disease as meaningful. Others note that the absolute difference on cognitive scales is small and may not be perceptible in daily life.
The GLP-1 receptor agonist connection adds another dimension. Several peptide drugs originally developed for diabetes (liraglutide, semaglutide, exenatide) have shown neuroprotective properties in preclinical Alzheimer's models, reducing amyloid-beta pathology and neuroinflammation through metabolic and anti-inflammatory mechanisms.[6] Clinical trials of GLP-1 agonists for Alzheimer's are underway. If they show benefit, it would suggest that targeting the metabolic and inflammatory environment may be as important as removing amyloid directly.
Does Amyloid-Beta Have Normal Functions?
A question that often gets lost in the Alzheimer's focus: does amyloid-beta do anything useful in a healthy brain?
Emerging evidence suggests yes. Amyloid-beta has been shown to have antimicrobial properties, acting as an innate immune peptide that traps and kills bacteria, fungi, and viruses. This "antimicrobial protection hypothesis" proposes that amyloid-beta production may be a defensive response to brain infections, and that Alzheimer's pathology represents an immune response that has spiraled out of control.[2]
At low concentrations, amyloid-beta may also participate in normal synaptic function and cholesterol transport. A 2026 study found that Abeta-40 levels in the bloodstream were associated with systemic metabolic health markers, suggesting the peptide may have functions beyond the brain.[7]
These findings raise a therapeutic concern: aggressive amyloid clearance might remove a peptide that has protective functions. This does not mean anti-amyloid therapy is wrong, but it suggests that the goal should be restoring balance rather than total elimination.
How the Brain Clears Amyloid-Beta
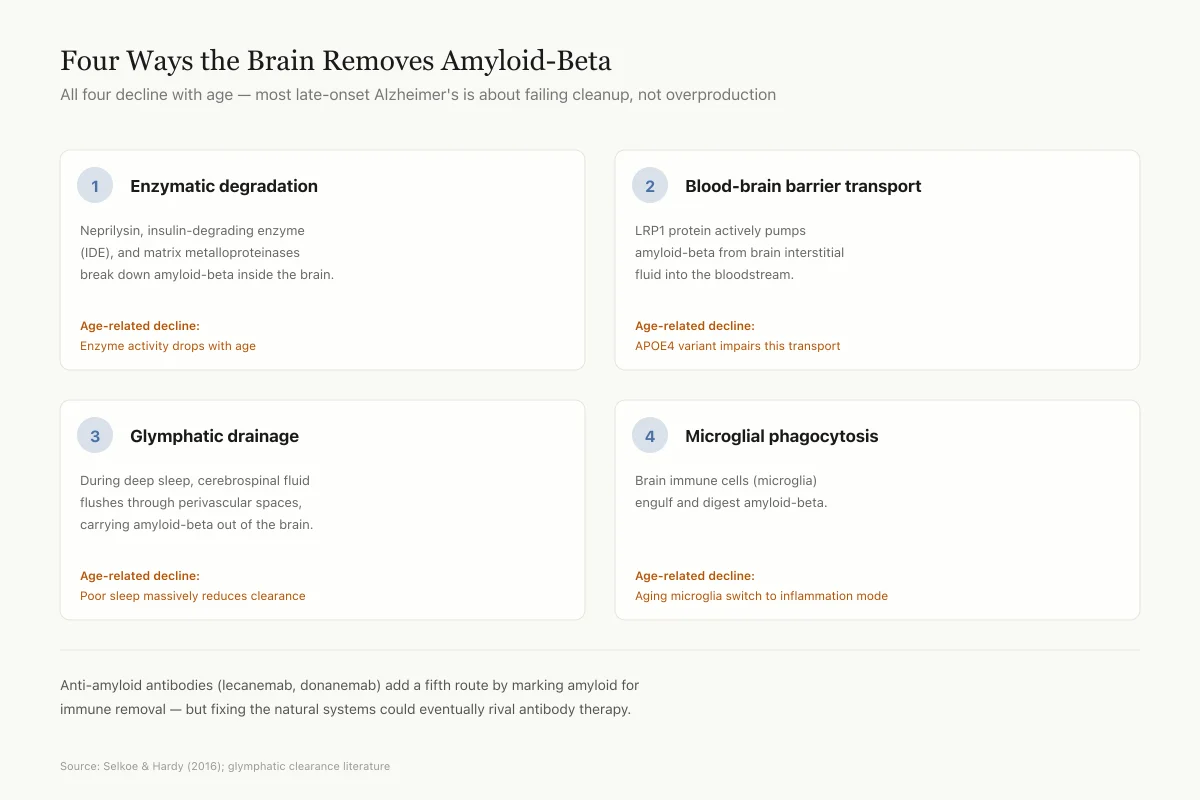
Understanding amyloid clearance is critical because most late-onset Alzheimer's cases result from impaired clearance rather than overproduction. The brain uses several mechanisms to remove amyloid-beta:
Enzymatic degradation: Enzymes including neprilysin, insulin-degrading enzyme (IDE), and matrix metalloproteinases break down amyloid-beta within the brain. Activity of these enzymes declines with age, contributing to gradual amyloid accumulation.[2]
Blood-brain barrier transport: Amyloid-beta is actively transported from the brain into the bloodstream by LRP1 (low-density lipoprotein receptor-related protein 1) on the blood side of the blood-brain barrier. APOE4 impairs this transport, which is one mechanism by which the APOE4 genotype increases Alzheimer's risk.
Glymphatic drainage: The brain's waste clearance system, the glymphatic pathway, flushes interstitial fluid (and dissolved amyloid-beta) through perivascular spaces during deep sleep. Sleep disruption substantially reduces glymphatic clearance, and epidemiological data consistently link poor sleep quality and duration with increased Alzheimer's risk. This may partly explain why sleep disturbances are both a risk factor for and an early symptom of Alzheimer's disease.
Microglial phagocytosis: Microglia (brain immune cells) engulf and degrade amyloid-beta. In early disease, this is protective. As the disease progresses, microglia become chronically activated and shift from a clearance phenotype to an inflammatory phenotype, producing cytokines that damage neurons while becoming less efficient at removing amyloid.
Each of these clearance mechanisms represents a potential therapeutic target independent of the antibody-based approaches currently in use. Enhancing natural clearance, rather than relying solely on exogenous antibodies to remove amyloid, could provide additional or alternative therapeutic strategies.
Clearance Pathways
Four Ways the Brain Removes Amyloid-Beta
All four decline with age — most late-onset Alzheimer's is about failing cleanup, not overproduction
Enzymatic degradation
Neprilysin, insulin-degrading enzyme (IDE), and matrix metalloproteinases break down amyloid-beta inside the brain.
Age-related decline: Enzyme activity drops with age
Blood-brain barrier transport
LRP1 protein actively pumps amyloid-beta from brain interstitial fluid into the bloodstream.
Age-related decline: APOE4 variant impairs this transport
Glymphatic drainage
During deep sleep, cerebrospinal fluid flushes through perivascular spaces, carrying amyloid-beta out of the brain.
Age-related decline: Poor sleep massively reduces clearance
Microglial phagocytosis
Brain immune cells (microglia) engulf and digest amyloid-beta.
Age-related decline: Aging microglia switch from cleanup to inflammation
Anti-amyloid antibodies (lecanemab, donanemab) add a fifth clearance route by marking amyloid for immune removal. But they're a costly, risky substitute for fixing the natural systems — enhancing sleep, supporting microglial function, or boosting transport could eventually rival antibody therapy.
Source: Selkoe & Hardy (2016); glymphatic clearance literature
 View as image
View as imageFuture Directions
The next phase of Alzheimer's peptide therapeutics is moving in several directions simultaneously. Combination therapies that target both amyloid and tau are in early clinical testing, based on the logic that amyloid initiates the disease but tau drives later progression. Anti-tau antibodies and tau aggregation inhibitors represent a parallel therapeutic track.
Prevention trials are testing whether anti-amyloid therapy can prevent or delay Alzheimer's in asymptomatic people with positive amyloid biomarkers. If amyloid is truly an early-stage trigger, treating it before symptoms appear could have a far greater impact than treating it after neurodegeneration is underway.
Blood-based biomarkers are making large-scale screening feasible for the first time. If plasma p-tau217 or Abeta-42/40 ratio tests can identify at-risk individuals years before symptoms, it opens the possibility of truly early intervention. The peptide at the heart of Alzheimer's may ultimately be targeted most effectively not after it has done its damage, but before the damage begins.
Limitations of the Evidence
Modest clinical benefit: Both lecanemab and donanemab slow decline rather than stop it. The absolute differences on cognitive scales are small. Whether patients perceive the benefit in daily life remains debated.
Safety risks: ARIA (brain swelling and microbleeds) affects 12-24% of patients. Most cases are asymptomatic, but deaths have occurred. Patients carrying the APOE4 gene (present in 60-75% of Alzheimer's patients) face higher ARIA risk.
Early-stage only: Both drugs were tested in early Alzheimer's. Their benefit in moderate or severe disease is unknown, and the tau data from donanemab suggest benefit diminishes as the disease advances.
Cost and access: Lecanemab requires biweekly intravenous infusions and regular MRI monitoring. Annual drug costs exceed $26,000 in the US. Many healthcare systems have limited coverage.
Causation versus correlation: Even with successful amyloid removal, the modest clinical benefit suggests amyloid may be one factor among many, not the sole driver. Tau pathology, neuroinflammation, vascular damage, and synaptic loss all contribute independently to cognitive decline.
Long-term data: Both trials lasted 18 months. Whether benefits accumulate, plateau, or diminish over years of treatment is unknown. Extension studies are ongoing.
The Bottom Line
Amyloid-beta is a 36-43 amino acid peptide fragment produced by enzymatic cleavage of APP in the brain. The amyloid cascade hypothesis, proposed in 1992, positions it as the initiating factor in Alzheimer's disease. After decades of failed trials, lecanemab and donanemab became the first drugs to demonstrate that amyloid removal can slow cognitive decline, validating amyloid as a therapeutic target. The benefit is modest (27-35% slowing of decline), the safety risks are real (ARIA in 12-24%), and the debate over whether amyloid removal alone is sufficient to meaningfully change the disease course continues.
Sources & References
- 1RPEP-07488·van Dyck, Christopher H et al. (2023). “Lecanemab in Early Alzheimer's Disease..” The New England journal of medicine.Study breakdown →PubMed →↩
- 2RPEP-03109·Selkoe, Dennis J et al. (2016). “The amyloid hypothesis of Alzheimer's disease at 25 years..” EMBO molecular medicine.Study breakdown →PubMed →↩
- 4RPEP-07393·Sims, John R et al. (2023). “Donanemab in Early Symptomatic Alzheimer Disease: The TRAILBLAZER-ALZ 2 Randomized Clinical Trial..” JAMA.Study breakdown →PubMed →↩
- 5RPEP-05468·Jeremic, Danko et al. (2021). “Past, present and future of therapeutic strategies against amyloid-β peptides in Alzheimer's disease: a systematic review..” Ageing research reviews.Study breakdown →PubMed →↩
- 6RPEP-03015·Li, Yanwei et al. (2016). “Diabetes Peptide Drugs Show Promise for Treating Alzheimer's and Parkinson's Disease.” Reviews in the neurosciences.Study breakdown →PubMed →↩
- 7RPEP-16153·Sopova, Kateryna et al. (2026). “Amyloid-beta (1-40) peptide is associated with systemic metabolic health..” European journal of clinical investigation.Study breakdown →PubMed →↩