Desmopressin: The Peptide for Diabetes Insipidus
Peptide Hormone Replacement Therapies
30+ years in clinical use
Desmopressin has been in continuous clinical use since the 1970s with an established safety profile across diabetes insipidus, bleeding disorders, and nocturnal enuresis.
Vande Walle et al., Current Drug Safety, 2007
Vande Walle et al., Current Drug Safety, 2007
If you only read one thing
Desmopressin is a slightly-tweaked copy of the natural hormone that tells your kidneys to hold on to water. Two small changes to the amino acid recipe made it about ten times stronger at concentrating urine and removed the side effect of raising blood pressure. It has been on the market since the 1970s and is considered a well-understood peptide drug. Doctors use it for three very different things: a condition called diabetes insipidus where people produce up to 20 liters of urine a day, certain mild bleeding disorders, and bedwetting in children. The main thing to watch is drinking too much water around a dose, which can drop blood sodium to dangerous levels.
Natural vasopressin (antidiuretic hormone, ADH) is a nine-amino-acid peptide produced by the hypothalamus and released from the posterior pituitary. It controls water balance by signaling the kidneys to concentrate urine. When vasopressin production or release fails, the result is central diabetes insipidus (DI): patients produce 3 to 20 liters of dilute urine daily, with unquenchable thirst and the constant risk of life-threatening dehydration. Desmopressin (1-deamino-8-D-arginine vasopressin, brand names DDAVP, Minirin, Stimate) solved this problem. Two strategic modifications to the natural vasopressin molecule, deamination at position 1 and substitution of D-arginine for L-arginine at position 8, created a peptide with dramatically enhanced antidiuretic potency, prolonged duration of action, and virtually no vasopressor (blood pressure-raising) activity.[2] Approved in the 1970s, desmopressin has been in continuous clinical use for over five decades, treating not only diabetes insipidus but also hemophilia A, von Willebrand disease, and nocturnal enuresis. This article covers its mechanism, clinical evidence, safety profile, and emerging delivery technologies. For related peptide hormone therapies, see the cluster articles on octreotide for carcinoid syndrome, pasireotide for Cushing's disease, and peptide treatment for acromegaly.
Key Takeaways
- Desmopressin has been in clinical use since the 1970s with a well-established safety profile; a 30-year safety review found hyponatremia as the primary risk, occurring mainly with improper fluid intake management (Vande Walle et al., Current Drug Safety, 2007).[1]
- The two amino acid modifications (deamination at position 1, D-arginine at position 8) increase antidiuretic potency 10-fold while eliminating the vasopressor activity that makes natural vasopressin unsuitable for chronic use.[2]
- Desmopressin treatment for von Willebrand disease has accumulated 30 years of clinical data, with Federici (2008) documenting its role as first-line treatment for type 1 VWD across surgical and emergency settings.[3]
- New V2 receptor agonist peptides with shorter half-lives are being developed for conditions where desmopressin's prolonged action creates hyponatremia risk (Wisniewski et al., J Med Chem, 2019).[4]
- GLP-1 receptor agonists may enhance desmopressin's effects in AVP-deficient patients, suggesting potential combination therapy (Nakhleh et al., 2024).[8]
- Microneedle patch delivery of desmopressin has shown enhanced transdermal absorption in preclinical studies, potentially offering a needle-free alternative to injection (Dashti et al., 2025).[9]
From vasopressin to desmopressin: two modifications that changed everything
Natural arginine vasopressin (AVP) is a terrible drug. Although it effectively treats diabetes insipidus in emergency settings, its action is too short (15 to 20 minutes intravenously), it raises blood pressure by constricting blood vessels through V1a receptor activation, and it is rapidly degraded by aminopeptidases. Chronic administration of native vasopressin for diabetes insipidus is impractical and dangerous.
Desmopressin was designed to fix all three problems through two molecular modifications. First, removal of the amino group from the cysteine at position 1 (deamination) makes the molecule resistant to aminopeptidase degradation, extending its half-life from minutes to hours. Second, substitution of D-arginine for L-arginine at position 8 shifts receptor selectivity dramatically toward V2 (the renal antidiuretic receptor) and away from V1a (the vascular vasopressor receptor). The resulting molecule is approximately 10 times more potent than vasopressin at inducing antidiuresis but has essentially no vasopressor effect at therapeutic doses.[2]
Glavas et al. (2022) reviewed the broader family of vasopressin analogs in a comprehensive assessment published in the European Journal of Medicinal Chemistry. Their analysis traced how systematic modifications to the vasopressin scaffold have produced peptides spanning the full spectrum of receptor selectivity, from V1a-selective agonists (for septic shock) to V2-selective agonists (desmopressin for antidiuresis) to mixed agonists and antagonists. Desmopressin remains the most clinically successful of all vasopressin analogs, in large part because the combination of V2 selectivity and metabolic stability was achieved with minimal structural change: just two modifications to a nine-amino-acid peptide.[5]
Two amino-acid changes
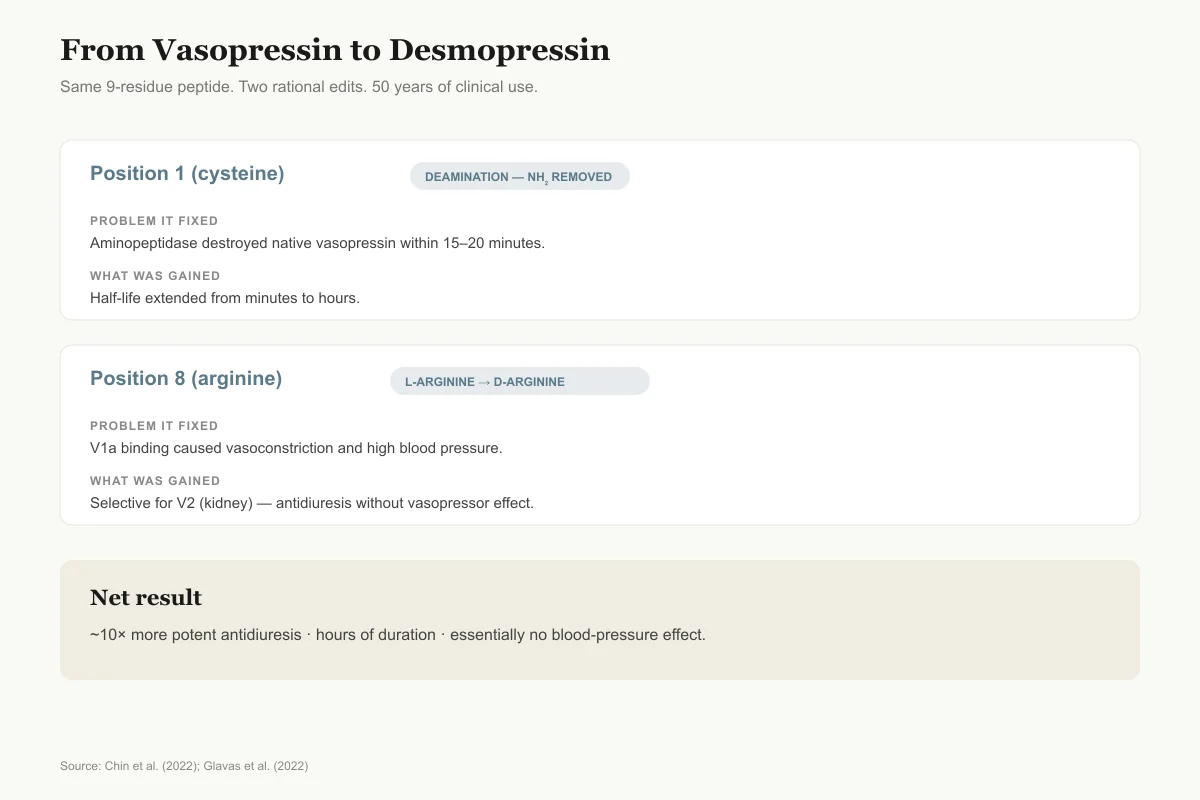
From Vasopressin to Desmopressin in Two Edits
Same 9-residue peptide. Two rational modifications. A drug that has held its position for 50 years.
Position 1 (cysteine)
Amino group removed (deamination)Problem it fixed
Aminopeptidase destroyed vasopressin within 15–20 minutes
What was gained
Half-life extended from minutes to hours
Position 8 (arginine)
L-arginine → D-arginineProblem it fixed
V1a receptor binding caused vasoconstriction and high blood pressure
What was gained
Selective for V2 (kidney) over V1a (vessels) — antidiuresis without vasopressor effect
Net result: ~10× more potent antidiuresis, hours of duration, essentially no blood-pressure effect.
Source: Chin et al. (2022); Glavas et al. (2022)
 View as image
View as imageMechanism: how desmopressin concentrates urine
Desmopressin's mechanism of action follows the natural vasopressin signaling cascade in the kidney collecting duct, amplified and prolonged.
The peptide binds V2 receptors on the basolateral membrane of principal cells in the renal collecting duct. This activates adenylyl cyclase, increasing intracellular cyclic AMP (cAMP). cAMP activates protein kinase A (PKA), which phosphorylates aquaporin-2 (AQP2) water channels stored in intracellular vesicles. Phosphorylated AQP2 molecules migrate to the apical (urine-facing) membrane and insert, creating water-permeable channels. Water from the dilute urine in the collecting duct flows down its osmotic gradient through AQP2 into the hyperosmolar renal medulla and back into the bloodstream.[2]
The result is concentrated urine and reduced urine volume. In patients with central DI who produce 10 or more liters of urine daily, a single dose of desmopressin can reduce output to 1 to 2 liters, eliminating the need for constant fluid intake and allowing normal sleep, work, and activity.
Chin et al. (2022) reviewed the pharmacological differences between desmopressin formulations in the European Journal of Clinical Pharmacology. Intranasal, oral, sublingual, and parenteral routes all deliver effective antidiuresis, but bioavailability varies dramatically: approximately 3 to 5% for oral tablets, 10% for intranasal spray, and 100% for subcutaneous/intravenous injection. Despite low oral bioavailability, the oral formulations provide reliable clinical effect because desmopressin is so potent that even the small fraction absorbed is sufficient for antidiuresis.[2]
Diabetes insipidus: the primary indication
Three clinical uses
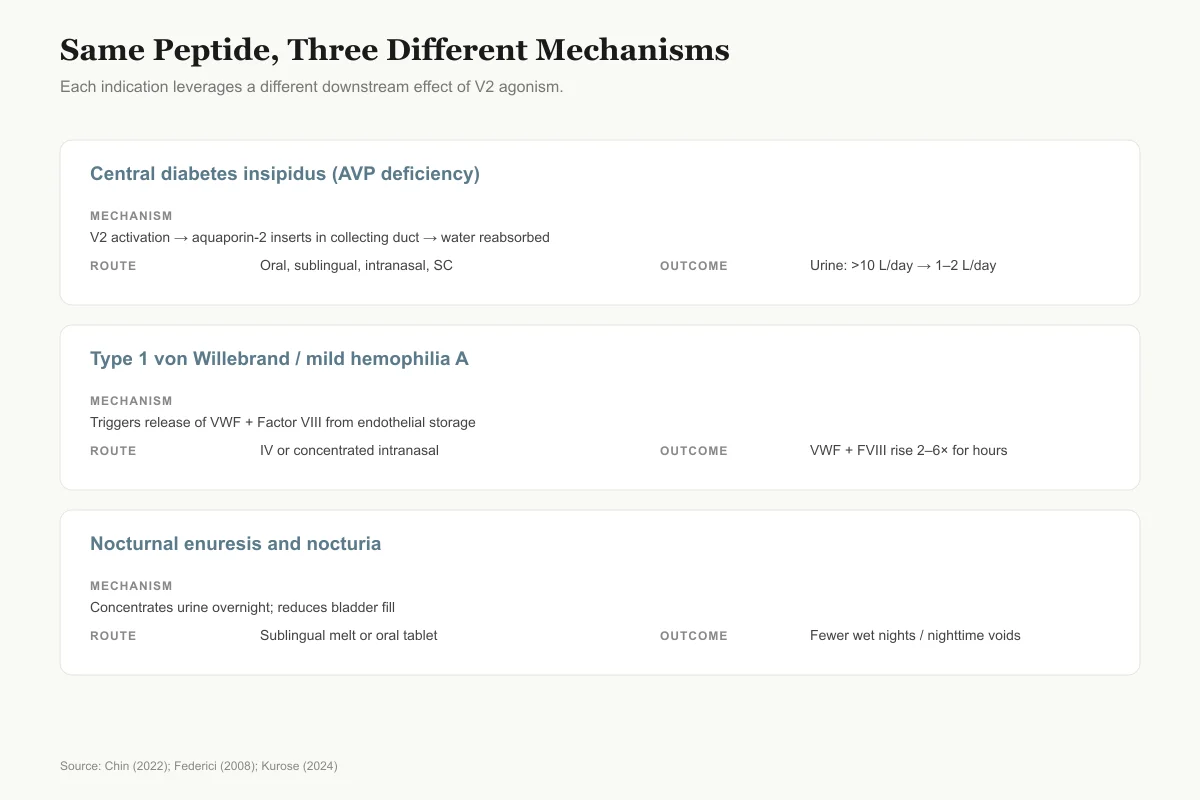
Same Peptide, Three Different Mechanisms
Each indication leverages a different downstream effect of V2 agonism.
Central diabetes insipidus (AVP deficiency)
Mechanism
V2 receptor activation → aquaporin-2 insertion in collecting duct
Route
Oral tablet, sublingual melt, intranasal, subcutaneous
Typical outcome
Urine volume falls from >10 L/day to 1–2 L/day
Type 1 von Willebrand disease / mild hemophilia A
Mechanism
Releases VWF and Factor VIII from endothelial Weibel-Palade bodies
Route
IV infusion or concentrated intranasal (Stimate)
Typical outcome
VWF and Factor VIII levels rise 2–6× for several hours
Nocturnal enuresis and nocturia
Mechanism
Concentrates urine overnight, shrinks bladder load
Route
Sublingual melt or oral tablet before bed
Typical outcome
Fewer wet nights in children; fewer nighttime voids in adults
Source: Chin (2022); Federici (2008); Kurose (2024)
 View as image
View as imageCentral diabetes insipidus results from insufficient vasopressin production or release, typically due to damage to the hypothalamus or posterior pituitary from surgery (especially transsphenoidal surgery for pituitary tumors), trauma, autoimmune destruction, genetic mutations, or infiltrative diseases. Without vasopressin, the kidneys cannot concentrate urine, and patients lose enormous volumes of water.
Chasseloup et al. (2024) reviewed the current understanding of diabetes insipidus (now renamed arginine vasopressin deficiency, AVP-D, to avoid confusion with diabetes mellitus). They noted that desmopressin remains the definitive treatment, converting a potentially lethal condition into a manageable chronic one. With appropriate desmopressin dosing, patients achieve normal urine concentration, normal serum osmolality, and can participate in all normal activities.[6]
Atila et al. (2024) expanded on the diagnosis and management of AVP deficiency, emphasizing that desmopressin dosing must be individualized. The balance between sufficient antidiuresis (preventing dehydration) and excessive antidiuresis (causing water retention and hyponatremia) requires careful titration, particularly during intercurrent illnesses when fluid intake and losses change unpredictably.[12]
Kleanthous et al. (2021) reported an unexpected complication when initiating DDAVP therapy in Wolfram syndrome patients. The first doses of desmopressin caused renal salt wasting due to elevated atrial natriuretic peptide levels, leading to potentially dangerous sodium losses. This case series illustrates that even a drug with decades of clinical experience can produce novel adverse effects in specific patient populations.[10]
Beyond diabetes insipidus: bleeding disorders
Desmopressin's second major clinical application was discovered serendipitously. When researchers noted that desmopressin infusion increased plasma levels of coagulation factor VIII and von Willebrand factor (VWF), they recognized its potential in hemostasis.
Federici (2008) published a comprehensive review of 30 years of desmopressin use in von Willebrand disease (1977 to 2007) in Haemophilia. The peptide stimulates release of VWF and factor VIII from endothelial cell storage pools (Weibel-Palade bodies), transiently raising plasma levels 2 to 6 times above baseline. This is sufficient to control mild to moderate bleeding in type 1 VWD (the most common form) and mild hemophilia A, often eliminating the need for blood product transfusion during minor surgical procedures, dental extractions, and menstrual management.[3]
Yankin et al. (2021) used impedance aggregometry to evaluate platelet function after DDAVP administration, providing evidence that desmopressin's hemostatic effects extend beyond factor level increases. The peptide appears to enhance platelet adhesion and aggregation, contributing to improved hemostasis through both coagulation factor release and direct platelet effects.[11]
The hemostatic indication highlights an important principle in peptide pharmacology: a peptide designed for one receptor interaction (V2-mediated antidiuresis) can have clinically valuable effects through secondary pathways (endothelial VWF release) that were not part of the original design. Desmopressin's hemostatic application was not predicted from its structure; it was discovered through careful clinical observation.
Nocturnal enuresis and nocturia
Desmopressin's ability to reduce nighttime urine production made it a natural candidate for treating nocturnal enuresis (bedwetting) in children and nocturia (excessive nighttime urination) in adults.
In pediatric nocturnal enuresis, desmopressin administered as a sublingual tablet or nasal spray before bedtime reduces overnight urine volume, allowing the bladder to hold urine until morning. Clinical trials have demonstrated significant reductions in wet nights per week, and the treatment has been in pediatric use for decades. The main safety concern is hyponatremia if children drink excessive fluids after taking the dose, which led to the withdrawal of the intranasal formulation for this indication in some countries while maintaining oral and sublingual formulations.
Kurose et al. (2024) evaluated the long-term effects of desmopressin treatment for nocturia in older adults. Nocturia is extremely common in the elderly (affecting over 50% of adults over 60) and significantly impacts sleep quality, fall risk, and quality of life. Desmopressin reduced nighttime voiding episodes, but the study also highlighted the increased hyponatremia risk in older patients, who have lower baseline sodium levels and less capacity to excrete water loads.[7]
Safety: 30 years of evidence
Hyponatremia risk stack
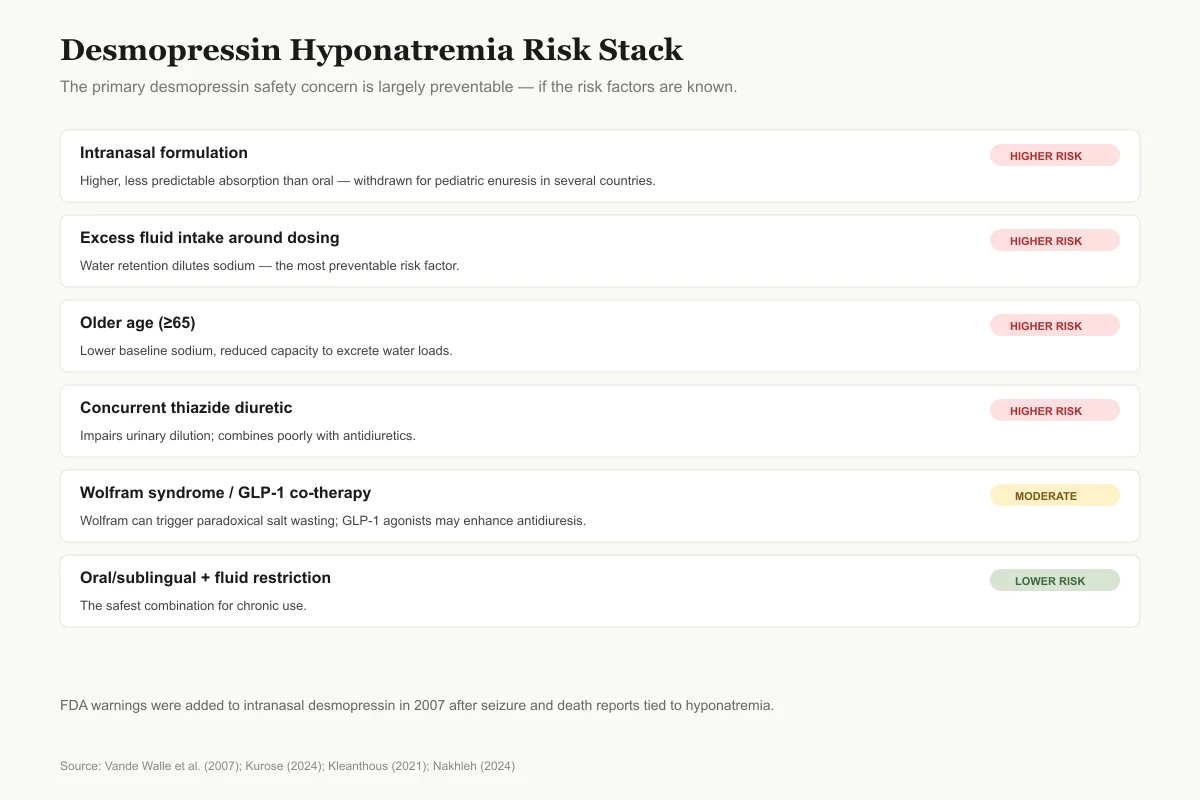
What Makes Low-Sodium Episodes More Likely
The primary desmopressin safety concern is largely preventable — if the risk factors are known.
Intranasal formulation
Higher riskHigher, less predictable absorption than oral — withdrawn for pediatric enuresis in several countries
Excess fluid intake around dosing
Higher riskWater retention dilutes sodium; most preventable risk factor
Older age (≥65)
Higher riskLower baseline sodium, reduced capacity to excrete water loads
Concurrent thiazide diuretic
Higher riskImpairs urinary dilution; combines poorly with antidiuretics
Wolfram syndrome
ModerateFirst doses can trigger paradoxical renal salt wasting via ANP (Kleanthous 2021)
Concurrent GLP-1 receptor agonist
ModerateMay enhance desmopressin effect in AVP-deficient patients (Nakhleh 2024)
Oral or sublingual formulation + fluid restriction
Lower riskThe safest combination for chronic use
FDA added warnings to intranasal desmopressin in 2007 after seizure and death reports tied to hyponatremia. Oral and sublingual use with fluid restriction has a considerably better safety profile.
Source: Vande Walle et al. (2007); Kurose (2024); Kleanthous (2021); Nakhleh (2024)
 View as image
View as imageVande Walle et al. (2007) published a comprehensive 30-year safety review of desmopressin in Current Drug Safety. The review confirmed that desmopressin is generally well tolerated across all indications, with hyponatremia (low blood sodium) as the primary safety concern.[1]
Hyponatremia occurs when desmopressin's antidiuretic effect causes water retention that dilutes serum sodium. The risk is highest when patients drink excessively after taking the dose, when the intranasal route is used (higher and less predictable absorption than oral), and in populations with low baseline sodium (elderly, patients on thiazide diuretics). Severe hyponatremia can cause seizures and, rarely, death. In 2007, the US FDA added warnings to intranasal desmopressin formulations after reports of seizures and two deaths associated with hyponatremia.
However, the safety review emphasized that hyponatremia is almost entirely preventable with proper fluid restriction. When patients are instructed to limit fluid intake for 1 hour before and 8 hours after desmopressin dosing, the risk drops dramatically. The oral and sublingual formulations, with their lower and more predictable bioavailability, have a better safety profile than intranasal formulations for the enuresis indication.
Beyond hyponatremia, desmopressin has remarkably few side effects. Headache, nausea, and nasal congestion (with intranasal use) occur but are generally mild. The absence of vasopressor activity means that unlike natural vasopressin, desmopressin does not raise blood pressure and can be used safely in patients with cardiovascular disease.[1]
Safety
ModerateLow blood sodium (hyponatremia) can be serious
Concern
Desmopressin's intended effect — water retention — can cause sodium levels to drop dangerously low if fluid intake is not limited. Severe hyponatremia can lead to seizures and, rarely, death. Risk is highest with intranasal dosing, in older adults, and when combined with thiazide diuretics.
What the research says
Restrict fluids for 1 hour before and up to 8 hours after dosing, use oral or sublingual formulations where possible, and monitor serum sodium during dose adjustments. In children treated for enuresis, do not use intranasal desmopressin.
Particularly relevant for: All desmopressin users, with highest concern in elderly and pediatric enuresis
What to do
Follow prescribed fluid-restriction instructions exactly. Report symptoms of hyponatremia (headache, nausea, confusion, weakness) to a clinician immediately.
Vande Walle et al. (2007), Current Drug Safety; FDA prescribing information
Emerging delivery technologies
Despite desmopressin's clinical success, its delivery remains suboptimal. Oral bioavailability is only 3 to 5%. Intranasal absorption is variable. Injectable administration requires clinic visits or patient self-injection training. These limitations have driven research into novel delivery platforms.
Dashti et al. (2025) developed polylactic acid microneedle patches for transdermal desmopressin delivery. Microneedles are tiny projections (typically 100 to 800 micrometers) that painlessly penetrate the outer skin layer and release drug into the dermis, bypassing the gastrointestinal degradation and first-pass metabolism that limit oral bioavailability. Their preclinical results showed enhanced transdermal absorption compared to passive skin permeation.[9]
Stie et al. (2022) took a different approach, developing mucoadhesive electrospun nanofiber-based hybrid systems for controlled desmopressin release. The nanofiber platform provides unidirectional drug release toward the mucosa while preventing loss into saliva or nasal fluid, potentially improving the consistency of sublingual or buccal desmopressin absorption.[13]
These delivery innovations address a broader challenge in peptide therapeutics: how to administer peptide drugs conveniently without injection while achieving reliable blood levels. Desmopressin, as one of the oldest and most widely used peptide drugs, serves as a test case for delivery technologies that could eventually be applied to newer peptide therapeutics.
Caliari et al. (2024) explored a complementary approach, studying the self-assembly behavior and cytocompatibility of desmopressin with sodium polystyrene sulfonate, investigating whether polymer-peptide interactions could improve formulation stability and bioavailability. Understanding how desmopressin interacts with excipients and formulation matrices at the molecular level may enable more rational design of delivery systems.[14]
Desmopressin as a model for peptide drug design
Desmopressin's five decades of clinical success offer broader lessons for peptide pharmacology that extend beyond its specific therapeutic indications.
The two-modification strategy demonstrates that minimal structural changes to a natural peptide hormone can dramatically alter pharmacological properties. Position 1 deamination addressed metabolic instability. Position 8 stereochemical inversion addressed receptor selectivity. Neither modification required extensive screening of combinatorial libraries or computational design; both were guided by understanding the relationship between peptide structure and the specific enzymatic and receptor interactions that needed to change. This rational approach, informed by deep mechanistic understanding rather than brute-force screening, remains a productive paradigm for peptide drug design.
The discovery of desmopressin's hemostatic application illustrates the importance of clinical observation in peptide drug development. The VWF-releasing effect was not predicted from the drug's molecular design. It emerged from careful measurement of coagulation parameters during clinical use for a different indication. This serendipitous discovery, which has benefited hundreds of thousands of patients with bleeding disorders, would have been missed in a purely target-based drug development program focused exclusively on V2 receptor pharmacology.
The safety lesson is equally instructive. Desmopressin's major adverse effect, hyponatremia, is a direct consequence of its intended pharmacology rather than an off-target toxicity. The same V2-mediated water retention that treats diabetes insipidus can, if uncontrolled, dilute serum sodium to dangerous levels. This pattern, where efficacy and toxicity are mechanistically linked, is common in peptide therapeutics and demands careful dose titration and patient education rather than molecular redesign.
Finally, desmopressin demonstrates that peptide drugs can achieve commercial viability and sustained clinical impact over decades. In an era when drug patent cliffs often limit the commercial lifespan of small molecules to 10 to 15 years, desmopressin's continued use across multiple indications and delivery formats provides a counterexample: peptide drugs with genuine clinical value can maintain relevance far beyond initial regulatory approval.
Next-generation vasopressin receptor agonists
Wisniewski et al. (2019) published in the Journal of Medicinal Chemistry on the discovery of potent, selective, and short-acting peptidic V2 receptor agonists. These new compounds were designed to address a specific clinical limitation of desmopressin: its long duration of action (6 to 24 hours depending on formulation), which extends the window during which hyponatremia can develop if fluid intake is not restricted.[4]
Short-acting V2 agonists would allow more precise temporal control of antidiuresis. For nocturia, a peptide that provides 4 to 6 hours of antidiuresis (covering the sleep period) and then wears off would enable normal morning fluid excretion, reducing hyponatremia risk. For procedural use in bleeding disorders, a short-acting agent could provide the hemostatic benefit of VWF release without prolonged antidiuresis afterward.
Nakhleh et al. (2024) reported a potentially important drug interaction: GLP-1 receptor agonists appeared to enhance the effects of desmopressin in individuals with AVP deficiency. With the explosive growth of GLP-1 agonist prescriptions for obesity and type 2 diabetes, this interaction is clinically relevant. Patients taking both desmopressin and a GLP-1 agonist may require dose adjustment to avoid excessive antidiuresis.[8]
What the evidence does not cover
Several gaps remain in the desmopressin evidence base despite five decades of clinical use.
Optimal dosing protocols for different DI etiologies have not been established through randomized trials. Most dosing is empirical, guided by clinical response and serum sodium monitoring. Whether patients with autoimmune-mediated DI respond differently than those with post-surgical DI, or whether genetic variants in the V2 receptor influence desmopressin sensitivity, has not been systematically studied.
The long-term renal effects of decades of daily desmopressin use in patients with childhood-onset DI are not well characterized. Patients diagnosed in childhood may use desmopressin for 60 or more years. Whether chronic V2 receptor stimulation affects kidney function, AQP2 expression, or medullary concentrating ability over these timescales is unknown.
Head-to-head comparisons of different desmopressin formulations (oral tablet, sublingual melt, intranasal spray, subcutaneous injection) in the same patient populations are sparse. Clinicians choose formulations based on convenience, cost, and perceived safety rather than comparative efficacy data from randomized trials.
The potential for desmopressin tachyphylaxis (reduced response with repeated dosing) in the hemostatic indication is recognized but incompletely understood. VWF stores in Weibel-Palade bodies can be depleted with repeated desmopressin doses, reducing the hemostatic response. Guidelines recommend limiting desmopressin use to one to three doses for bleeding events, but the kinetics of VWF store replenishment vary between individuals and are not predictable.
The global accessibility of desmopressin also warrants attention. In high-income countries, desmopressin is widely available and affordable. In low- and middle-income countries, where diabetes insipidus goes undiagnosed or untreated far more frequently, access is inconsistent. Generic desmopressin formulations exist, but cold chain requirements for some preparations, cost of monitoring serum sodium, and lack of specialist endocrine care create barriers to appropriate use. Given that untreated central DI can be fatal from dehydration, particularly in young children and the elderly, improving global access to this established peptide therapy remains a public health priority.
Whether emerging oral peptide technologies could further improve desmopressin's already-existing oral formulation is an open question. Current oral desmopressin tablets achieve clinical efficacy despite only 3 to 5% bioavailability. Permeation enhancers, enteric coatings, and other oral peptide delivery strategies developed for GLP-1 agonists and other newer peptide drugs could theoretically improve desmopressin's oral absorption, potentially allowing lower doses that maintain efficacy while further reducing hyponatremia risk. However, the economic case for reformulating a generic, inexpensive drug with decades of satisfactory clinical performance may not justify the development costs.
The Bottom Line
Desmopressin is a two-modification peptide masterpiece: deamination at position 1 and D-arginine substitution at position 8 converted the natural hormone vasopressin into a selective, stable, long-acting antidiuretic drug with minimal vasopressor activity. Over five decades of clinical use across diabetes insipidus, bleeding disorders, and nocturnal enuresis have established its efficacy and safety profile, with hyponatremia as the primary risk, manageable through fluid restriction. Emerging delivery technologies (microneedle patches, nanofiber systems) and next-generation short-acting V2 agonists are addressing the remaining limitations of this foundational peptide therapeutic.
Sources & References
- 1RPEP-01299·Vande Walle, Johan et al. (2007). “Desmopressin 30 years in clinical use: a safety review..” Current drug safety.Study breakdown →PubMed →↩
- 2RPEP-06049·Chin, Xinyi et al. (2022). “Desmopressin therapy in children and adults: pharmacological considerations and clinical implications..” European journal of clinical pharmacology.Study breakdown →PubMed →↩
- 3RPEP-01337·Federici, A B (2008). “The use of desmopressin in von Willebrand disease: the experience of the first 30 years (1977-2007)..” Haemophilia : the official journal of the World Federation of Hemophilia.Study breakdown →PubMed →↩
- 4RPEP-04555·Wiśniewski, Kazimierz et al. (2019). “Discovery of Potent, Selective, and Short-Acting Peptidic V2 Receptor Agonists..” Journal of medicinal chemistry.Study breakdown →PubMed →↩
- 5RPEP-06143·Glavaš, Mladena et al. (2022). “Vasopressin and Its Analogues: From Natural Hormones to Multitasking Peptides..” International journal of molecular sciences.Study breakdown →PubMed →↩
- 6RPEP-07965·Chasseloup, Fanny et al. (2024). “Diabetes Insipidus Explained: Why Vasopressin Deficiency Treatment Doesn't Always Restore Quality of Life.” Annales d'endocrinologie.Study breakdown →PubMed →↩
- 7RPEP-08613·Kurose, Hirofumi et al. (2024). “Long-Term Desmopressin Treatment Is Effective and Safe for Nighttime Urination in Older Men.” International journal of urology : official journal of the Japanese Urological Association.Study breakdown →PubMed →↩
- 8RPEP-08931·Nakhleh, Afif et al. (2024). “GLP-1 Drugs May Require Desmopressin Dose Cuts in Diabetes Insipidus Patients.” Pituitary.Study breakdown →PubMed →↩
- 9RPEP-10624·Dashti, Amirhossein et al. (2025). “Development of polylactic acid microneedles for enhanced transdermal delivery of desmopressin peptides: A computational study..” Journal of pharmaceutical sciences.Study breakdown →PubMed →↩
- 10RPEP-05504·Kleanthous, Kleanthis et al. (2021). “Starting DDAVP Causes Dangerous Salt Loss via 50-Fold ANP Surge in Wolfram Syndrome.” Indian journal of pediatrics.Study breakdown →PubMed →↩
- 11RPEP-05905·Yankin, Igor et al. (2021). “The use of impedance aggregometry to evaluate platelet function after the administration of DDAVP in healthy dogs treated with aspirin or clopidogrel..” American journal of veterinary research.Study breakdown →PubMed →↩
- 12RPEP-07782·Atila, Cihan et al. (2024). “Understanding Arginine Vasopressin Deficiency: How It's Diagnosed, Treated, and Why Oxytocin Matters Too.” Nature reviews. Endocrinology.Study breakdown →PubMed →↩
- 13RPEP-06516·Stie, Mai Bay et al. (2022). “Mucoadhesive Electrospun Nanofiber-Based Hybrid System with Controlled and Unidirectional Release of Desmopressin..” International journal of molecular sciences.Study breakdown →PubMed →↩
- 14RPEP-07918·Caliari, Ana B et al. (2024). “Desmopressin Combined with a Polymer Forms Nanostructures That Selectively Inhibit Aggressive Breast Cancer Cells.” Soft matter.Study breakdown →PubMed →↩