Antiviral Peptides Against Influenza
Antiviral Peptides and Influenza
Femtomolar potency
Bovine lactoferrin C-lobe fragments inhibited influenza virus hemagglutination at femtomolar concentrations across H1N1, H3N2, H5N1, and H7N1 subtypes.
Ammendolia et al., Pathogens and Global Health, 2012
Ammendolia et al., Pathogens and Global Health, 2012
If you only read one thing
Tamiflu, the main prescription flu drug, targets one viral protein — and resistance pops up regularly. Antiviral peptides attack the flu virus through four different routes at once: ripping open the viral shell, blocking the hook it uses to grab your cells, gluing its fusion machinery together, and turning up your body's interferon alarm. Some of these peptides come from frog skin, some from human immune cells, and some are engineered from antibody fragments. A frog peptide called urumin protected mice from lethal flu, even from drug-resistant strains. None are FDA-approved yet — the main hurdles are keeping peptides intact in the respiratory tract long enough to work, and proving dose-for-dose efficacy in humans. Several intranasal programs are in preclinical development.
Influenza viruses kill 290,000 to 650,000 people worldwide each year, and the current antiviral arsenal is thin. Oseltamivir (Tamiflu) and zanamivir target a single viral protein, neuraminidase, and resistance mutations emerge regularly in circulating strains. Antiviral peptides offer a fundamentally different approach. These molecules, many derived from the same host defense peptides that protect organisms from bacteria, can target influenza through multiple mechanisms simultaneously: blocking the hemagglutinin protein that the virus uses to enter cells, disrupting the viral lipid envelope, and activating innate immune defenses. Some act at femtomolar concentrations. Others work against drug-resistant strains that have rendered conventional antivirals ineffective. This article maps the full evidence landscape of antiviral peptides against influenza, from endogenous human peptides to synthetic fusion inhibitors.
Key Takeaways
- Flu kills up to 650,000 people worldwide every year, and the main drug (Tamiflu) targets just one viral protein. Resistance keeps popping up.
- Antiviral peptides attack the flu virus through four different routes at once — far harder for the virus to evolve around than a single-target pill.
- Some peptides from frog skin protected mice from lethal flu, even from drug-resistant strains. One fragment works at concentrations so low it's hard to measure.
- Your own body makes a flu-fighting peptide called LL-37. In mice, it reduced flu severity about as much as a prescription antiviral — by tearing open the virus itself.
- None of these peptides are FDA-approved yet. The main problem is keeping them intact in the nose and throat long enough to work against flu.
- Lactoferrin supplements sold for "flu protection" don't work the way the in vitro data suggests. Swallowed lactoferrin gets digested before reaching where flu infects.
- The realistic future isn't a pill — it's an intranasal peptide spray, and several are in preclinical development right now.
How influenza enters cells and why it matters for peptide design
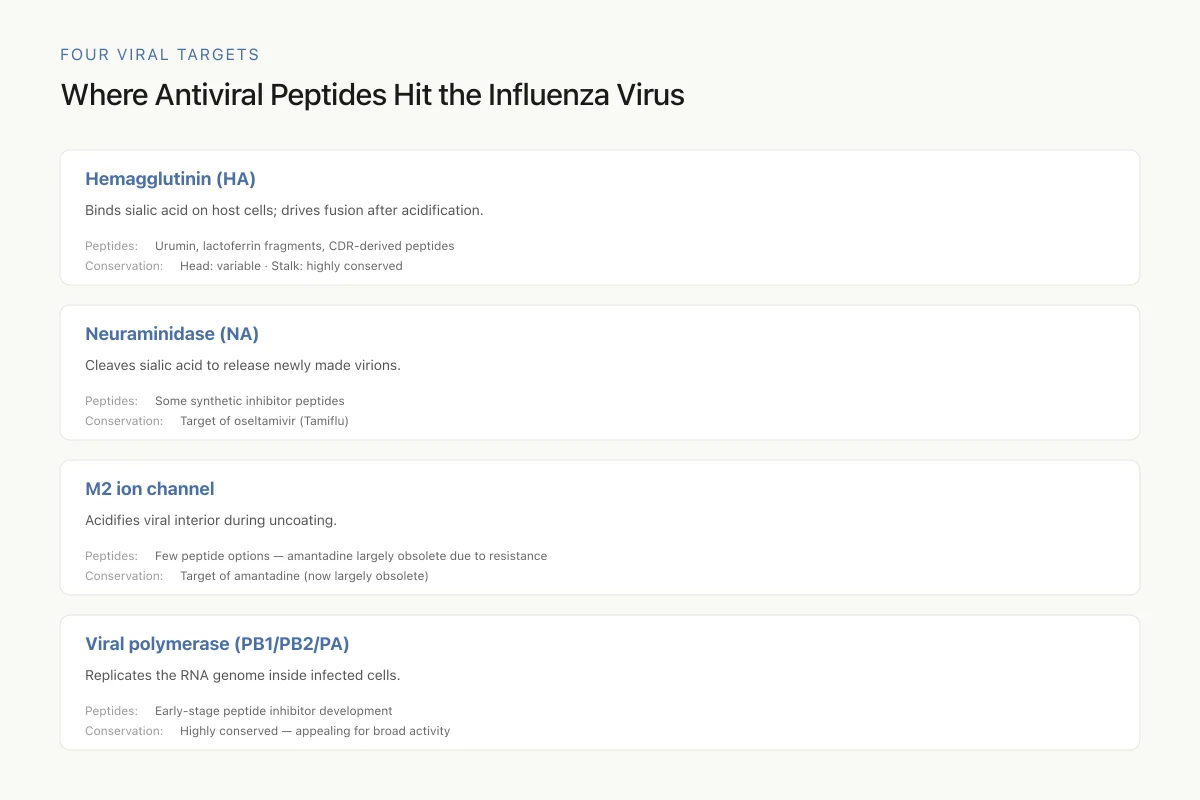
Influenza infection begins when the viral hemagglutinin (HA) protein binds sialic acid residues on the surface of respiratory epithelial cells. The virus is then internalized into endosomes, where the acidic environment triggers a massive conformational change in HA. The HA2 subunit, which contains a highly conserved fusion peptide, unfolds and inserts into the host cell membrane, pulling the viral and cellular membranes together until they fuse. The viral RNA genome then enters the cytoplasm to begin replication.
Each step in this process represents a potential target for antiviral peptides. Peptides can block the initial HA-sialic acid binding, prevent the pH-triggered conformational change, inhibit the fusion peptide insertion, or disrupt the viral envelope before cell contact occurs. The HA stem region, where the fusion machinery resides, is particularly attractive because it is highly conserved across influenza subtypes, meaning a peptide that binds there could work against H1N1, H3N2, H5N1, and other strains that differ substantially in their HA head domains.[1]
Beyond HA, three other viral proteins serve as peptide targets. Neuraminidase (NA) cleaves sialic acid to release newly formed virions from infected cells. The M2 ion channel acidifies the viral interior during uncoating. The viral polymerase complex (PB1, PB2, PA) replicates the RNA genome. Peptides have been developed against all four targets, though the largest body of research focuses on HA inhibition.[1]
Four Viral Targets
Where Antiviral Peptides Hit the Influenza Virus
Tamiflu targets one protein (neuraminidase). Drug resistance happens when that protein mutates. Peptides targeting multiple viral proteins simultaneously — or targeting conserved regions that can't mutate without killing the virus — are much harder to outrun.
Source: Agamennone et al. (2022); Holthausen et al. (2017)
 View as image
View as imageLL-37: how your own cathelicidin fights the flu
The human cathelicidin LL-37 is a 37-residue peptide produced by neutrophils, macrophages, and epithelial cells during infection. It is one of the most studied endogenous antiviral peptides against influenza.
Barlow et al. (2011) demonstrated that LL-37 reduced influenza disease severity in infected mice to a degree comparable to zanamivir, the prescription neuraminidase inhibitor. The peptide showed dose-dependent antiviral activity and, when administered prophylactically, significantly reduced viral titers in the lungs. LL-37 acted as a direct virucidal agent, verified through plaque reduction assays, rather than solely through immunomodulation.[3]
Tripathi et al. (2013) dissected LL-37's mechanism and found it was fundamentally different from other innate immune molecules that fight influenza. Unlike surfactant protein D, which inhibits viral binding to cell surface receptors, LL-37 did not prevent viral association with epithelial cells. Unlike human neutrophil defensins, it did not aggregate viral particles. Instead, LL-37 appeared to directly disrupt the viral lipid envelope, compromising viral integrity before cell entry could occur. This unique mechanism means LL-37 could theoretically complement, rather than duplicate, the action of other antiviral molecules in the respiratory tract.[4]
More recent work has revealed additional dimensions of LL-37's antiviral role. Cerps et al. (2025) showed that LL-37 increased rhinovirus-induced interferon-beta expression in human airway epithelial cells through TLR3-dependent signaling. While this study focused on rhinovirus rather than influenza, it demonstrated that LL-37 enhances the innate antiviral interferon response in the airways, an effect that would benefit defense against influenza as well.[11]
Safety
ModerateLL-37's immune-boosting is a double-edged sword in newborns
Concern
The same cathelicidin that helps adult airways fight flu is actively harmful in neonatal mice. Rao et al. (2025) found that the mouse cathelicidin CRAMP drove excessive inflammation in infant influenza infection, worsening outcomes rather than improving them.
What the research says
Peptide immunomodulators interact with immune state and age in complex ways. What protects one population can damage another. Adult-focused antiviral peptides would need separate safety work before being used in pediatric or neonatal patients.
Particularly relevant for: Neonates, immunocompromised patients, anyone with hyperinflammatory conditions
What to do
The current LL-37 evidence supports its role as an endogenous defense — not as an administered drug at any age, and especially not in infants until more data exists.
Rao et al. (2025), pediatric influenza cathelicidin study
The complexity of LL-37's role in infection was underscored by Rao et al. (2025), who found that the murine cathelicidin CRAMP (the mouse equivalent of LL-37) was actually toxic during neonatal influenza infection. In neonatal mice, excessive cathelicidin-driven inflammation worsened disease outcomes rather than improving them, suggesting that the peptide's immunomodulatory effects can be a double-edged sword depending on age and inflammatory context.[13]
This finding has broader implications for antiviral peptide development. Any peptide that modulates immune function, in addition to directly killing viruses, carries the risk of excessive inflammation in certain patient populations. The neonatal immune system's heightened sensitivity to inflammatory signals is an extreme case, but it illustrates why in vivo safety testing across multiple age groups and immune states will be critical before LL-37 or its derivatives could be used as administered therapeutics against influenza.
Urumin: a frog peptide that destroys drug-resistant flu
Urumin emerged from an unexpected source: the skin secretions of the South Indian frog Hydrophylax bahuvistara. Amphibian skin peptides have long been recognized as a rich source of antimicrobial molecules, but Holthausen et al. (2017) identified something rarer: a peptide with potent and specific anti-influenza activity.
Urumin is a 27-amino acid cationic peptide that physically destroys influenza virions bearing H1 hemagglutinin. Using electron microscopy, the researchers visualized urumin literally ripping apart viral particles. The peptide binds the conserved stalk region of H1 HA, a domain that drug-resistant influenza strains cannot easily mutate without losing the ability to fuse with host cell membranes. This stalk-targeting mechanism means urumin remained active against H1N1 strains resistant to oseltamivir and amantadine.[2]
In mouse experiments, intranasal urumin administration protected animals from lethal influenza challenge. The peptide's selectivity was notable: it was virucidal against H1-bearing influenza viruses but showed minimal activity against H3N2 strains, indicating that it targets a specific structural feature of the H1 hemagglutinin stalk rather than acting as a nonspecific membrane disruptor.[2]
The subtype specificity that makes urumin so effective against H1 also limits its scope. A pandemic H3N2 strain would likely be resistant. However, urumin serves as a proof of concept that peptides can achieve high-potency, mechanism-specific antiviral activity by targeting conserved HA structural domains. The same rational approach, identifying conserved stalk epitopes specific to other HA subtypes, could yield peptides tailored to H3, H5, or H7 hemagglutinins. A cocktail of subtype-specific peptides might achieve the broad coverage of a universal antiviral while retaining the high potency that comes from precise structural targeting.
Urumin's discovery also illustrates why amphibian skin secretions remain a productive source of bioactive peptides. Frogs co-exist with a vast microbial and viral environment, and their skin peptide repertoire has been shaped by millions of years of selection pressure. Screening these natural peptide libraries against human pathogens has yielded candidates that no computational design approach predicted.
Lactoferrin-derived peptides: broad-spectrum influenza inhibition
Lactoferrin, a glycoprotein abundant in mammalian milk, tears, and respiratory secretions, has been studied for antiviral activity since the 1990s. Its peptide fragments show some of the most potent anti-influenza activity recorded for any natural compound.
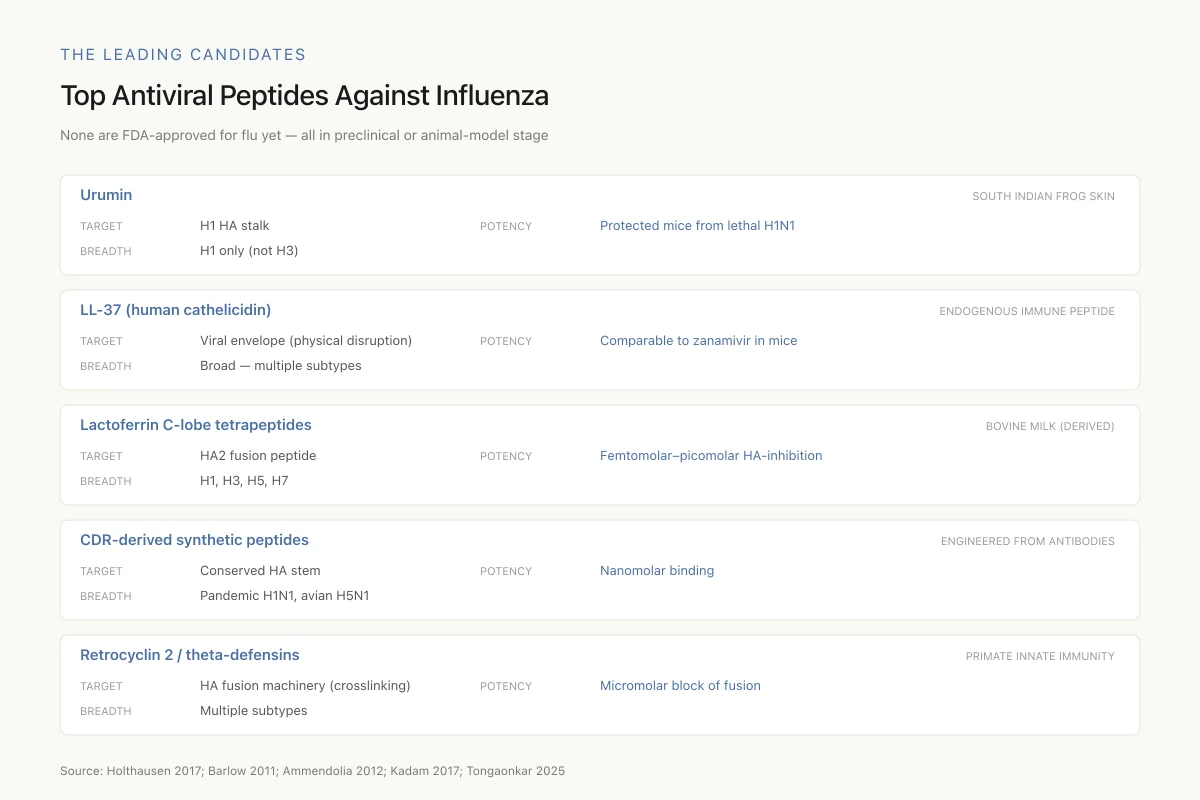
The Leading Candidates
Top Antiviral Peptides Against Influenza
None are FDA-approved for flu yet — all in preclinical or animal-model stage
The field faces a classic trade-off: urumin is hyper-potent but narrow; lactoferrin peptides are broad but their in vivo potency hasn't been matched to their in vitro numbers. CDR-derived synthetic peptides are the most drug-like candidates, shrinking antibody breadth into peptide-sized molecules.
Source: Holthausen 2017; Barlow 2011; Ammendolia 2012; Kadam 2017; Tongaonkar 2025
 View as image
View as imageAmmendolia et al. (2012) dissected bovine lactoferrin into its N-lobe and C-lobe domains and tested each against multiple influenza subtypes. All anti-influenza activity mapped to the C-lobe. This fragment bound the HA2 region of hemagglutinin, the highly conserved domain containing the fusion peptide, and inhibited both hemagglutination and cell infection across H1N1, H3N2, H5N1, and H7N1 subtypes. The inhibitory concentrations were remarkable: femtomolar to nanomolar range in hemagglutination inhibition assays.[6]
Scala et al. (2017) identified three specific tetrapeptides derived from the bovine lactoferrin C-lobe that retained broad anti-influenza activity. These four-amino-acid sequences inhibited viral infection at femtomolar to picomolar concentrations. The tiny size of these peptides, just four residues, makes them attractive starting points for drug development because they can be synthesized cheaply, are relatively resistant to proteolysis, and can potentially be modified for improved pharmacokinetics.[7]
The broad-spectrum activity of lactoferrin peptides contrasts sharply with urumin's subtype specificity. By targeting the universally conserved fusion peptide region of HA2, lactoferrin fragments achieve cross-subtype activity, though the translation of femtomolar hemagglutination inhibition to in vivo efficacy remains incompletely characterized. The hemagglutination inhibition assay, while standard in the field, measures peptide binding to free HA protein rather than to HA embedded in intact virions within a mucus-covered respiratory epithelium. The gap between assay potency and therapeutic potency may be substantial, and in vivo dose-response studies in animal models are needed to calibrate expectations.
The practical advantage of tetrapeptide inhibitors is their simplicity. At four amino acids, these molecules are far smaller than conventional protein therapeutics, can be produced by straightforward solid-phase synthesis, and may be amenable to oral or intranasal formulation. Their small size also reduces the likelihood of immunogenicity, a concern with larger peptide therapeutics.
Synthetic peptide inhibitors of hemagglutinin fusion
While natural host defense peptides provided early leads, synthetic design has produced some of the most potent anti-influenza peptides reported.
Kadam et al. (2017) published a landmark study in Science describing synthetic peptides designed from the complementarity-determining region (CDR) loops of broadly neutralizing human antibodies FI6v3 and CR9114. These antibodies bind the conserved HA stem epitope and neutralize multiple influenza subtypes. By extracting the critical binding loops and engineering them as standalone peptides, the researchers created molecules with nanomolar affinity for the HA stem. The optimized peptides neutralized influenza A group 1 viruses, including 2009 pandemic H1N1 and avian H5N1 strains, by blocking the pH-triggered conformational rearrangements required for membrane fusion.[5]
This work represented a significant conceptual advance: it demonstrated that the binding specificity of a large antibody (approximately 150 kDa) could be miniaturized into a peptide of approximately 3 to 4 kDa while retaining nanomolar potency. The size reduction is roughly 40-fold, yet the critical molecular contacts with the HA stem epitope are preserved. Peptides of this size are far cheaper to manufacture than monoclonal antibodies and potentially amenable to intranasal delivery, which would place the drug directly at the site of influenza infection. The structural data also provided atomic-resolution maps of peptide-HA interactions, enabling iterative optimization of binding affinity and pharmacokinetic properties through rational modification of non-binding residues.
Gonepudi et al. (2025) extended this structure-guided approach to design peptide inhibitors targeting class I viral fusion proteins more broadly. Using computational modeling of the pre-fusion to post-fusion HA conformational transition, they identified peptide sequences that could stabilize the pre-fusion state, preventing the structural rearrangement needed for membrane fusion. This approach may be generalizable beyond influenza to other enveloped viruses that use class I fusion proteins, including paramyxoviruses and coronaviruses.[10]
Meng et al. (2025) took a dual-targeting approach, designing a branched peptide that simultaneously blocked influenza virus and rhinovirus entry. The peptide targeted both viral factors and host cell membrane components involved in the fusion process, achieving broad antiviral activity against respiratory viruses through a single molecule. This strategy addresses a clinical reality: respiratory infections often involve co-infections or sequential infections with multiple viruses.[8]
Defensins and theta-defensins: innate immune peptides with antiviral roles
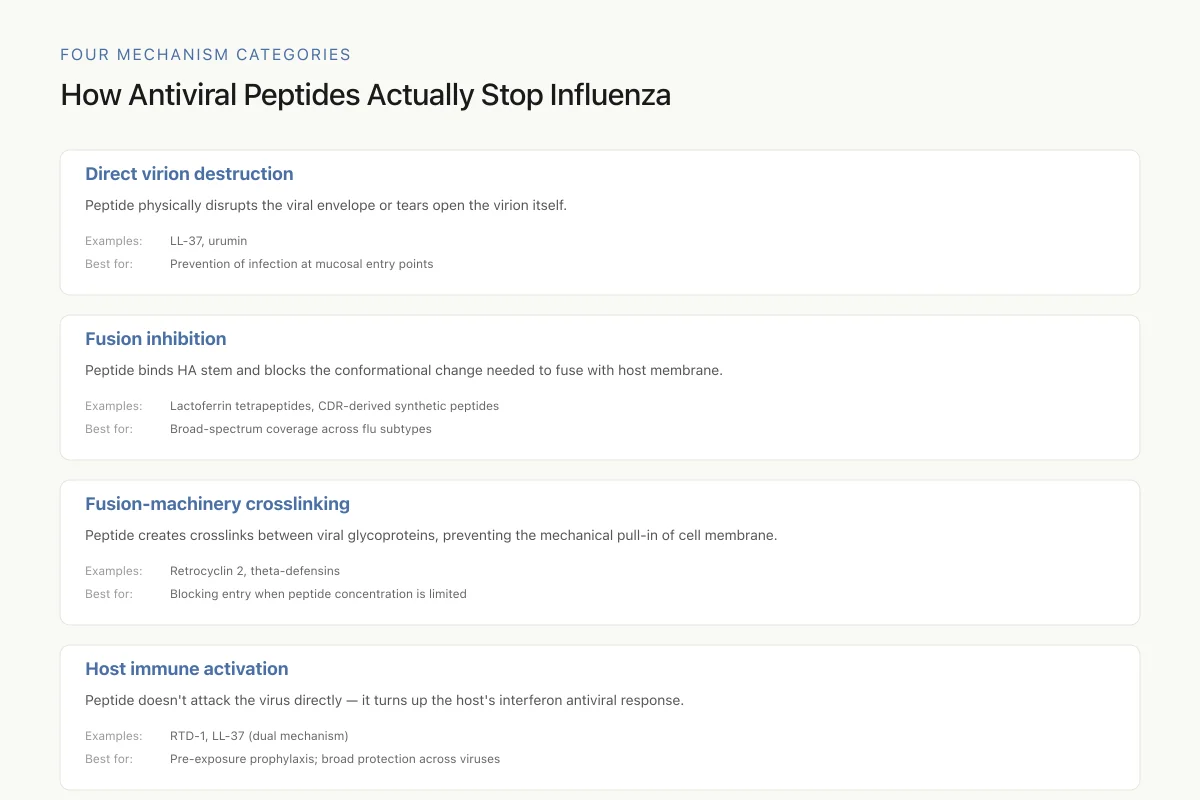
Four Mechanism Categories
How Antiviral Peptides Actually Stop Influenza
The most promising therapeutic approach combines at least two of these. A peptide that both physically wrecks virions and boosts interferon would handle both ongoing and future infections. Most current single-peptide candidates do only one.
Source: Agamennone et al. (2022); Tripathi et al. (2013); Tongaonkar et al. (2025)
 View as image
View as imageAlpha-defensins, released by neutrophils during innate immune responses, contribute to antiviral defense through mechanisms distinct from both LL-37 and lactoferrin. Retrocyclin 2, a theta-defensin, blocks influenza entry by interacting with surface glycoproteins and producing crosslinks that prevent hemagglutinin-mediated membrane fusion. Unlike LL-37, which disrupts the viral envelope, retrocyclin interferes with the fusion machinery itself.[1]
Tongaonkar et al. (2025) demonstrated that rhesus theta-defensin 1 (RTD-1), a macrocyclic peptide, activated interferon and antiviral signaling pathways in human monocytes. Rather than acting directly on the virus, RTD-1 enhanced the host cell's own antiviral defenses by upregulating interferon-stimulated genes. This immunomodulatory mechanism provides protection that is inherently broad-spectrum, since interferon responses are effective against diverse viral families.[9]
The distinction between direct antiviral activity (LL-37 disrupting viral membranes, urumin destroying virions, lactoferrin blocking HA fusion) and indirect immunomodulatory activity (theta-defensins activating interferon pathways) is central to understanding how peptides fight influenza. Direct antivirals must be present at sufficient concentration at the time of viral exposure, while immunomodulators can prime defenses before infection occurs. The most effective therapeutic strategy may involve peptides that combine both properties, or cocktails of peptides with complementary mechanisms that address different stages of the viral life cycle.
The macrocyclic structure of theta-defensins like RTD-1 is worth noting in the context of drug development. Cyclic peptides are inherently more resistant to protease degradation than their linear counterparts, addressing one of the key pharmacokinetic barriers that limits peptide therapeutics. RTD-1's ability to activate interferon responses, combined with its natural stability, makes it a structurally privileged scaffold for antiviral peptide development.
Peptide-based influenza vaccines
Antiviral peptides are not limited to direct viral inhibition. Self-assembling peptide nanostructures are being developed as platforms for influenza vaccine delivery.
Roe et al. (2026) demonstrated that presenting hemagglutinin antigens on self-assembling peptide nanofibers bearing built-in adjuvants increased heterologous immune responses against influenza. The peptide scaffold served dual roles: it organized HA antigens in a multivalent display that enhanced B cell recognition, and it provided intrinsic adjuvant activity that stimulated robust immune responses without the need for additional chemical adjuvants. The nanofiber format generated cross-reactive antibodies against influenza strains not included in the vaccine formulation, addressing a key limitation of current seasonal flu vaccines.[12]
This application illustrates how peptide science extends beyond direct antiviral activity into immunology and vaccine design, creating multiple routes from peptide research to clinical impact against influenza.
Current limitations and the path to clinical use
Despite impressive in vitro and animal data, no antiviral peptide has yet been approved specifically for influenza treatment. Several barriers explain this gap.
Serum stability remains the primary pharmacokinetic challenge. Unmodified peptides are rapidly degraded by proteases in the respiratory tract and bloodstream. Lactoferrin-derived tetrapeptides show femtomolar activity in cell-free assays, but achieving comparable concentrations in the respiratory epithelium of a living patient requires either chemical modifications (D-amino acid substitution, cyclization, PEGylation) or local delivery strategies (intranasal administration, inhalation). Each approach introduces new considerations around manufacturing cost, formulation stability, and regulatory requirements.
The disconnect between in vitro potency and in vivo efficacy is common across the field. Urumin protected mice from lethal influenza at doses that would be challenging to scale to human lungs. Lactoferrin fragments that inhibit hemagglutination at femtomolar concentrations may bind serum proteins, reducing free drug available at the site of infection. These pharmacokinetic uncertainties require systematic in vivo dose-finding studies that many promising peptide candidates have not yet received.
Subtype specificity is both an advantage and a limitation. Urumin's H1-specific activity makes it useless against H3N2 epidemics. Lactoferrin's broad-spectrum activity may come at the cost of lower potency against individual subtypes compared to optimized strain-specific peptides. The ideal therapeutic would combine broad-spectrum coverage with high potency, a goal that remains elusive.
Timing of administration matters more for antiviral peptides than for many other therapeutics. Peptides that block viral entry are most effective when present before or during the initial hours of infection. Once influenza has established productive replication in respiratory epithelial cells, peptides targeting HA-mediated entry have a diminished impact. This constraint favors prophylactic or very early therapeutic use, potentially in outbreak settings or for high-risk contacts, rather than treatment of established infection.
The competitive landscape has shifted. Monoclonal antibodies targeting the HA stem (like the broadly neutralizing antibodies that inspired Kadam's peptide designs) have advanced further in clinical development. Peptides must demonstrate clear advantages in cost, delivery convenience (intranasal vs. intravenous), or breadth of coverage to justify development alongside antibody-based approaches. The cost advantage is real: peptide synthesis is orders of magnitude cheaper than monoclonal antibody production, and the intranasal delivery route that peptides are suited for places the drug directly at the mucosal surface where influenza initiates infection.
The Bottom Line
Antiviral peptides target influenza through at least four distinct mechanisms: direct virion destruction (urumin, LL-37), hemagglutinin fusion inhibition (lactoferrin fragments, synthetic CDR-derived peptides), host immune activation (theta-defensins), and vaccine antigen presentation (self-assembling peptide nanofibers). The evidence spans potencies from femtomolar to micromolar, with activity demonstrated across H1N1, H3N2, H5N1, and drug-resistant strains. The primary unresolved challenge is translating this in vitro and animal model evidence into pharmacokinetically viable human therapeutics.
Sources & References
- 1RPEP-05962·Agamennone, Mariangela et al. (2022). “Antiviral Peptides as Anti-Influenza Agents..” International journal of molecular sciences.Study breakdown →PubMed →↩
- 2RPEP-03320·Holthausen, David J et al. (2017). “An Amphibian Host Defense Peptide Is Virucidal for Human H1 Hemagglutinin-Bearing Influenza Viruses..” Immunity.Study breakdown →PubMed →↩
- 3RPEP-01736·Barlow, Peter G et al. (2011). “Antiviral activity and increased host defense against influenza infection elicited by the human cathelicidin LL-37..” PloS one.Study breakdown →PubMed →↩
- 4RPEP-02295·Tripathi, Shweta et al. (2013). “The human cathelicidin LL-37 inhibits influenza A viruses through a mechanism distinct from that of surfactant protein D or defensins..” The Journal of general virology.Study breakdown →PubMed →↩
- 5RPEP-03336·Kadam, Rameshwar U et al. (2017). “Potent peptidic fusion inhibitors of influenza virus..” Science (New York.Study breakdown →PubMed →↩
- 6RPEP-01895·Ammendolia, Maria Grazia et al. (2012). “Milk Protein Fragments Blocked Influenza Virus at Incredibly Low Concentrations.” Pathogens and global health.Study breakdown →PubMed →↩
- 7RPEP-03457·Scala, Maria Carmina et al. (2017). “Lactoferrin-derived Peptides Active towards Influenza: Identification of Three Potent Tetrapeptide Inhibitors..” Scientific reports.Study breakdown →PubMed →↩
- 8RPEP-12539·Meng, Xinjie et al. (2025). “A branched peptide targets virus and host to block influenza virus and rhinovirus entry..” Antimicrobial agents and chemotherapy.Study breakdown →PubMed →↩
- 9RPEP-13821·Tongaonkar, Prasad et al. (2025). “Cyclic defensin peptide RTD-1 activates interferon antiviral pathways and blocks SARS-CoV-2 infection in human cells.” Journal of leukocyte biology.Study breakdown →PubMed →↩
- 10RPEP-11144·Gonepudi, Narendra Kumar et al. (2025). “Structure-Guided Design of Peptide Inhibitors Targeting Class I Viral Fusion Proteins..” Pathogens (Basel.Study breakdown →PubMed →↩
- 11RPEP-10332·Cerps, Samuel et al. (2025). “LL-37 Boosts Your Airway's Antiviral Response to the Common Cold Virus.” Biochemistry and biophysics reports.Study breakdown →PubMed →↩
- 12RPEP-16004·Roe, Emily F et al. (2026). “Self-Assembling Peptide Nanofiber Flu Vaccines Generate Antibodies Effective Against 30 Years of Viral Drift.” Acta biomaterialia.Study breakdown →PubMed →↩
- 13RPEP-13201·Rao, Abhishek S et al. (2025). “Cathelicidin-related antimicrobial peptide (CRAMP) is toxic during neonatal murine influenza virus infection..” Journal of immunology (Baltimore.Study breakdown →PubMed →↩