Antimicrobial Peptides vs Antibiotics: The Evidence
Antimicrobial Peptides
5,099 AMPs Cataloged
The APD3 database now catalogs 5,099 antimicrobial peptides from bacteria, plants, fungi, and animals. These molecules kill pathogens through mechanisms fundamentally different from conventional antibiotics.
APD3 Database, University of Nebraska Medical Center, 2025
APD3 Database, University of Nebraska Medical Center, 2025
If you only read one thing
Antimicrobial peptides are tiny natural defenders that every living thing — from frogs to humans — uses to kill bacteria. They work by physically punching holes in bacterial membranes instead of jamming a single molecular target like regular antibiotics do. That makes them far harder for bacteria to outsmart. After decades of research, only a handful are actually in use (mostly topical), because the same peptide that kills bacteria can also damage human cells at high doses, and peptides get chewed up fast in blood. The likely future isn't replacing antibiotics — it's using AMPs alongside lower-dose antibiotics to stretch the ones we still have.
Antimicrobial resistance killed an estimated 1.27 million people directly in 2019 and was associated with 4.95 million deaths globally, according to the Global Burden of Disease study published in The Lancet. Conventional antibiotics target specific bacterial proteins or enzymes, and bacteria evolve resistance to these single targets with alarming speed. Antimicrobial peptides (AMPs) attack bacterial membranes through physical disruption, a mechanism that has persisted in nature for hundreds of millions of years with limited resistance evolution.[1] Whether AMPs can translate from ancient biological defense into modern therapeutics is the central question of this field. The evidence is substantial but complicated.
Key Takeaways
- Antimicrobial peptides are tiny natural defenders every living thing — frogs, humans, plants — uses to kill bacteria.
- They don't work like regular antibiotics. Instead of jamming a single molecular target, they physically punch holes in bacterial membranes.
- That makes them far harder for bacteria to evolve around. The mechanism has worked in nature for hundreds of millions of years with limited resistance.
- Scientists have cataloged over 5,000 of them. Only a handful have reached actual medical use, and those are mostly topical creams and gels.
- Antibiotic resistance killed 1.27 million people in 2019. These peptides are one of the most actively researched ways to attack that problem.
- The main obstacles: peptides get chewed up in blood within minutes, and the same membrane-attack that kills bacteria can damage human cells.
- The likely future isn't replacing antibiotics. It's using these peptides alongside antibiotics to stretch the effective life of the drugs we still have.
The Scale of the Antibiotic Resistance Crisis
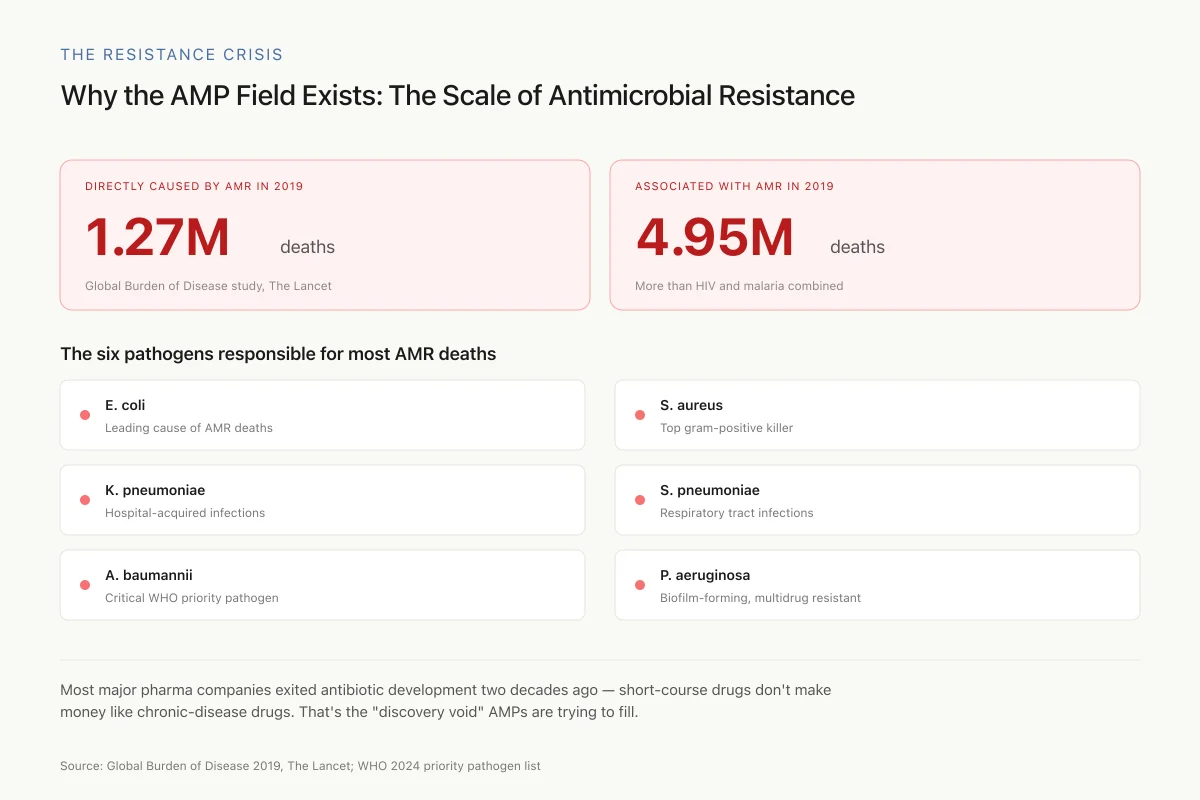
The urgency behind AMP research is driven by numbers. The 2019 Global Burden of Antimicrobial Resistance study, published in The Lancet, attributed 1.27 million deaths directly to bacterial antimicrobial resistance and found it associated with 4.95 million deaths globally in a single year. Six pathogens (E. coli, S. aureus, K. pneumoniae, S. pneumoniae, A. baumannii, and P. aeruginosa) were responsible for the majority of resistance-associated deaths. The WHO's 2024 updated priority pathogen list classified several of these as critical-priority threats requiring urgent new therapeutic strategies.[10]
The Resistance Crisis
Why the AMP Field Exists: The Scale of Antimicrobial Resistance
Most major pharma companies exited antibiotic development two decades ago — short-course drugs don't make money like chronic-disease drugs do. That's the "discovery void" AMPs are trying to fill, alongside bacteriophage therapy and anti-virulence strategies.
Source: Global Burden of Disease 2019, The Lancet; WHO 2024 priority pathogen list
 View as image
View as imageThe pipeline of new conventional antibiotics is thin. Most large pharmaceutical companies exited antibiotic development over the past two decades because short-course treatments generate less revenue than chronic disease drugs, and resistance can render a new antibiotic obsolete within years. This economic reality has created what infectious disease researchers call the "discovery void": a widening gap between the pace of resistance evolution and the pace of new drug development. AMPs represent one of the most active attempts to fill this void, alongside bacteriophage therapy, anti-virulence strategies, and microbiome-based interventions.
What Are Antimicrobial Peptides?
Antimicrobial peptides are short chains of amino acids, typically 12 to 50 residues long, that organisms produce as part of their innate immune defense. They are found in virtually every kingdom of life: bacteria produce bacteriocins, plants produce defensins and NCR peptides, insects produce cecropins, amphibians produce magainins, and mammals produce cathelicidins and defensins.[2] As of January 2025, the APD3 database catalogs 5,099 peptides, including 3,306 natural AMPs from six kingdoms: 410 bacterial bacteriocins, 5 from archaea, 8 from protists, 29 from fungi, 268 from plants, and 2,580 from animals, alongside 1,299 synthetic and 231 computationally predicted peptides.
Most AMPs share three properties that enable their antimicrobial function. They are cationic (positively charged), which attracts them to negatively charged bacterial membranes. They are amphipathic, meaning they have both water-loving and fat-loving regions, allowing them to insert into lipid bilayers. And they are small enough to reach the bacterial membrane before proteases can degrade them. The selectivity of AMPs for bacterial over mammalian membranes arises from a fundamental difference in membrane composition: bacterial membranes are rich in negatively charged phospholipids (phosphatidylglycerol, cardiolipin), while mammalian cell membranes have a neutral outer leaflet dominated by phosphatidylcholine and cholesterol.[1]
The human body produces its own arsenal. Alpha-defensins are stored in neutrophil granules at concentrations high enough to kill bacteria on contact. The cathelicidin LL-37 is expressed in epithelial cells, macrophages, and neutrophils across the skin, lungs, and gastrointestinal tract. Beta-defensins line epithelial surfaces throughout the body. These peptides do not merely supplement the immune system; in many contexts, they are the immune system's first line of killing.
AMPs are classified structurally into four main groups: alpha-helical peptides (like LL-37 and magainins, which form helical structures upon membrane contact), beta-sheet peptides (like defensins, stabilized by disulfide bonds), extended peptides (rich in specific amino acids like proline, tryptophan, or histidine), and loop peptides (containing a single disulfide bond creating a loop structure).[2] Each structural class interacts with bacterial membranes differently, contributing to the diversity of killing mechanisms.
For a broader look at how nature's organisms produce antimicrobial compounds, see Amphibian Skin Peptides: The Pharmacy on a Frog's Back.
How AMPs Kill Bacteria: Three Membrane Models
The mechanisms by which AMPs destroy bacteria have been studied for decades. Kim Brogden's 2005 review in Nature Reviews Microbiology established the framework that the field still uses, identifying three primary models of membrane disruption plus several intracellular mechanisms.[1]
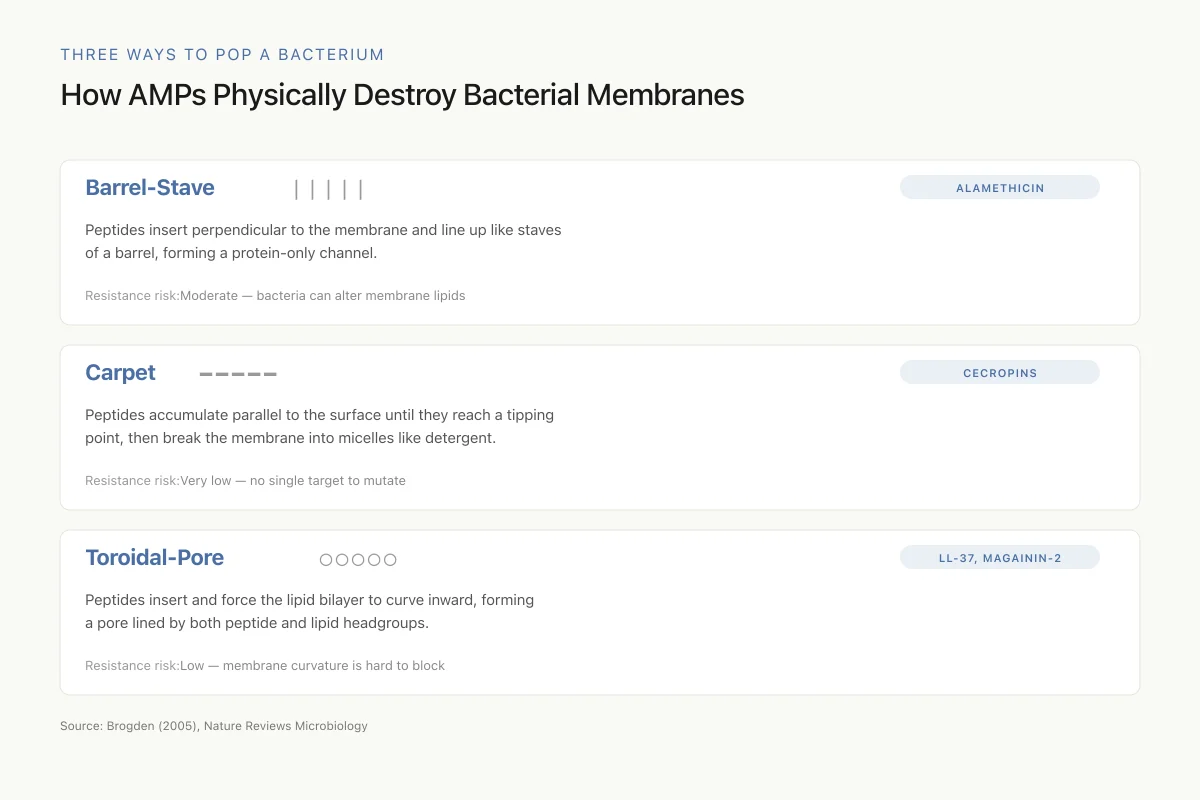
Three Ways to Pop a Bacterium
How AMPs Physically Destroy Bacterial Membranes
Conventional antibiotics target a protein. A single mutation in that protein can produce resistance. AMPs attack the lipid membrane itself — a physical structure bacteria can't redesign without dying. That's the core argument for why AMPs age more gracefully than antibiotics.
Source: Brogden (2005), Nature Reviews Microbiology
 View as image
View as imageBarrel-Stave Model
AMP molecules insert perpendicularly into the lipid bilayer and oligomerize to form a barrel-like channel. The hydrophobic faces of the peptides align with the lipid core; the hydrophilic faces line the interior of the pore. The result is a transmembrane channel that allows uncontrolled ion flux and cytoplasmic leakage. Alamethicin is the best-characterized peptide operating through this mechanism.
Carpet Model
Rather than inserting into the membrane, peptides accumulate on the bacterial surface in a carpet-like fashion, lying parallel to the membrane. When a threshold concentration is reached, the accumulated peptides disrupt the membrane in a detergent-like manner, breaking it into micelles. This model does not require specific peptide-peptide interactions or pore formation, which is why bacteria find it difficult to develop resistance: there is no single molecular target to mutate.
Toroidal-Pore Model
Peptides insert into the membrane and cause the lipid monolayers to bend continuously, forming a pore lined by both peptide molecules and lipid headgroups. Unlike barrel-stave pores where only peptides line the channel, toroidal pores involve the membrane itself curving inward. Magainin-2, one of the first AMPs discovered in frog skin, and the human cathelicidin LL-37 operate through variations of this model.
Beyond Membrane Disruption
Brogden's review also noted that membrane pore formation may not be the only killing mechanism. Subsequent research confirmed that many AMPs, after crossing the membrane, target intracellular processes: inhibiting DNA replication, RNA transcription, protein synthesis, and enzymatic activity.[1] A 2026 study demonstrated this dual functionality directly by designing FPON, the first antimicrobial peptide engineered to both disrupt bacterial membranes and inhibit protein translation simultaneously.[3] FPON specifically targeted gram-negative bacteria, exhibited low toxicity to mammalian cells, and showed significant activity against drug-resistant bacteria both in vitro and in mouse infection models. For more on how AMPs interact with specific drug-resistant organisms, see AMPs Against MRSA: Peptide Approaches to Drug-Resistant Staph.
Why AMPs Resist Resistance (Mostly)
The central argument for AMPs as antibiotic alternatives is that bacteria struggle to evolve resistance against them. The reasoning is straightforward: conventional antibiotics typically target a single protein (a ribosomal subunit, a cell wall enzyme, a DNA gyrase), and a single point mutation in that protein can confer resistance. AMPs target the bacterial membrane itself, a fundamental physical structure that bacteria cannot easily redesign without losing viability.[2]
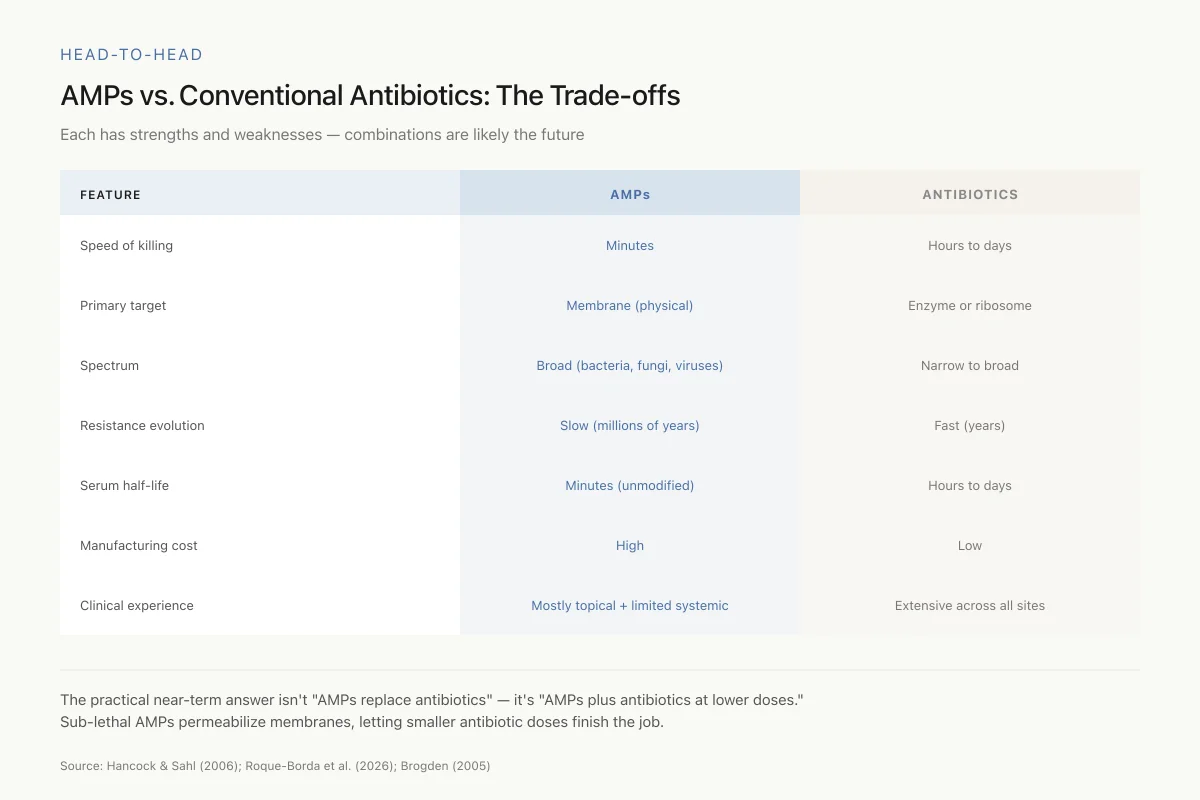
Head-to-Head
AMPs vs. Conventional Antibiotics: The Trade-offs
Each has strengths and weaknesses — which is why combinations are likely the future
The most practical near-term answer isn't "AMPs replace antibiotics" — it's "AMPs plus antibiotics at lower doses." Sub-lethal doses of AMPs can permeabilize bacterial membranes, letting smaller amounts of conventional antibiotics finish the job. That extends the lifespan of existing drugs.
Source: Hancock & Sahl (2006); Roque-Borda et al. (2026); Brogden (2005)
 View as image
View as imageHancock and Sahl's 2006 analysis in Nature Biotechnology noted that AMPs have coexisted with bacteria for hundreds of millions of years, yet widespread high-level resistance remains rare.[4] This contrasts sharply with conventional antibiotics, where clinically significant resistance often emerges within years of a drug's introduction.
But "rare" is not "absent." Cheung et al. demonstrated in 2018 that Staphylococcus aureus uses the Pmt ABC transporter to actively pump out human antimicrobial peptides.[5] Their study provided the first direct in vivo evidence that AMP resistance contributes to actual infection: S. aureus strains with functional Pmt caused more severe skin infections in mice, and this virulence advantage disappeared when AMP activity was experimentally removed from the system. The implication is clear: AMP resistance mechanisms already exist in clinically important pathogens and already matter during human infections.
Other bacterial resistance strategies include modifying membrane lipid composition to reduce negative charge, producing proteases that degrade AMPs before they reach the membrane, and secreting molecules that sequester AMPs in the extracellular space. These mechanisms tend to provide partial, low-level resistance rather than the complete resistance often seen with conventional antibiotics, but they are real and should inform therapeutic AMP design. For a deeper analysis of how bacteria develop and deploy these defenses, see Can Bacteria Become Resistant to Antimicrobial Peptides?.
Two Therapeutic Classes: Killers and Modulators
Hancock and Sahl's framework divided AMPs into two distinct therapeutic categories with different clinical trajectories.[4]
Direct Antimicrobial Peptides
These kill bacteria through the membrane disruption and intracellular mechanisms described above. They are rapid-acting (killing within minutes rather than hours), broad-spectrum, and potent. Clinical trials have tested several direct-acting AMPs including pexiganan (a magainin derivative), omiganan (a cathelicidin-derived synthetic), and various defensin analogs. To date, clinical success has been limited primarily to topical applications.[4]
A 2022 study advanced the topical delivery approach by encapsulating omiganan in liposomes for treating atopic dermatitis and psoriasis.[6] The liposomal formulation achieved 72% encapsulation efficiency with vesicles of 120 nm diameter, demonstrated controlled release, and showed better skin permeation than conventional gel formulations. In mouse models of atopic dermatitis, omiganan liposomal gel significantly reduced inflammatory markers.
Immunomodulatory Host-Defense Peptides
These peptides boost the host's own infection-fighting capacity rather than killing bacteria directly. They modulate innate immune responses: recruiting immune cells to infection sites, enhancing phagocytosis, promoting wound healing, and dampening excessive inflammatory responses that cause tissue damage during sepsis. The theta-defensin RTD-1 illustrates this dual capacity: a 2025 study showed it simultaneously activates antiviral interferon pathways through JAK-STAT1 signaling while inhibiting pro-inflammatory NF-kB signaling.[7] RTD-1 also inhibited SARS-CoV-2 infection in human cell models.
This immunomodulatory approach may ultimately prove more clinically viable than direct killing because it works with the immune system rather than trying to replicate what antibiotics do.
AMPs Against Biofilms
Biofilms represent one of the most intractable problems in infectious disease. Bacteria embedded in biofilm communities can tolerate antibiotic concentrations 100 to 1,000 times higher than planktonic (free-floating) bacteria. AMPs show particular promise in this area because their membrane-targeting mechanism can penetrate biofilm matrices that block conventional antibiotics.
A 2026 study characterized NCR169C17-38, a plant-derived AMP fragment, and found it effectively inhibited biofilm formation and eradicated pre-formed biofilms of Acinetobacter baumannii at sub-MBC concentration (3.2 micromolar), a result not observed for the conventional antimicrobial peptide polymyxin B.[8] Transcriptomic analysis revealed a global shutdown of bacterial metabolic functions, with 450 of 503 differentially expressed genes being downregulated. This multi-target assault on bacterial function makes resistance evolution substantially harder than against drugs with single targets.
A complementary strategy emerged from biofilm disruption research. Kurbatfinski et al. (2025) showed that bacteria newly released from biofilms by a targeted monoclonal antibody become transiently hypersensitive to killing by respiratory tract AMPs including beta-defensin 1, beta-defensin 3, and LL-37.[9] In three animal models of biofilm infections, antibody-mediated disruption alone (without co-delivered antibiotics) achieved rapid bacterial clearance and disease resolution. This two-step approach, breaking the biofilm shield and letting innate AMPs clean up, could transform management of chronic infections like recurrent sinusitis, otitis media, and device-related infections. For the full landscape of AMP-biofilm interactions, see AMPs and Biofilm Infections: Breaking Through Bacterial Fortresses.
Synergy: AMPs Plus Conventional Antibiotics
Using AMPs alongside existing antibiotics rather than as replacements may be the most practical near-term clinical application. A 2026 comprehensive review in Pharmacological Reviews cataloged dozens of successful AMP-antibiotic synergistic combinations against WHO priority pathogens.[10]
The mechanistic basis for synergy is logical: AMPs disrupt bacterial membranes, creating entry points for antibiotics that would otherwise be excluded. Several specific mechanisms drive synergistic effects:
- Membrane permeabilization: AMPs create pores or weaken membrane integrity, allowing antibiotics to reach intracellular targets they could not otherwise access.
- Efflux pump inhibition: Some AMPs interfere with bacterial efflux pumps that normally expel antibiotics, restoring antibiotic sensitivity.
- Biofilm penetration: AMPs penetrate biofilm matrices, delivering antibiotics into communities that would otherwise be protected.
- Intracellular delivery: AMPs that translocate across membranes can carry co-administered antibiotics into the bacterial cytoplasm.
The practical consequence is that sub-inhibitory concentrations of AMPs, doses too low to kill bacteria on their own, can reduce the effective antibiotic dose needed by several fold. This approach could extend the clinical lifespan of existing antibiotics while reducing dose-related toxicity. For detailed analysis of specific AMP-antibiotic pairings and their clinical potential, see Combination Therapy: AMPs Plus Antibiotics for Synergistic Effects.
LL-37 Derivatives: A Case Study in AMP Optimization
Human cathelicidin LL-37 is the most studied human AMP and illustrates both the potential and the challenges of therapeutic AMP development. It is 37 amino acids long, broad-spectrum against gram-positive and gram-negative bacteria, and has immunomodulatory properties. It is also expensive to synthesize, susceptible to proteolytic degradation, and toxic to mammalian cells at higher concentrations.
A 2025 study designed a rationally optimized LL-37 derivative that addressed several of these limitations.[11] The derivative showed lower minimum inhibitory concentrations (MICs) and faster bactericidal kinetics than the parent LL-37 against clinical multidrug-resistant E. coli and ESKAPE pathogens (Enterococcus faecium, S. aureus, Klebsiella pneumoniae, A. baumannii, Pseudomonas aeruginosa, Enterobacter spp.). It maintained low cytotoxicity to human epithelial and immune cells at concentrations exceeding bactericidal levels. ELISA quantification showed immunomodulatory effects on IL-6 and TNF-alpha at bactericidal concentrations, confirming that optimized derivatives can retain the dual antimicrobial-immunomodulatory properties of the parent peptide.
Machine Learning Is Accelerating AMP Discovery
Traditional AMP discovery relied on isolating peptides from natural sources, a slow and labor-intensive process. Machine learning has fundamentally changed this. A 2024 review in Nature Reviews Bioengineering documented how ML models now predict antimicrobial activity, hemolytic toxicity, and proteolytic stability from peptide sequences alone.[12]
The approach works because AMP activity depends on physicochemical properties (charge, hydrophobicity, amphipathicity, secondary structure) that are computationally predictable from amino acid sequences. ML models trained on databases of known AMPs can screen millions of candidate sequences in hours, identifying novel peptides with predicted activity profiles that would have taken years to discover experimentally.
Several recent advances illustrate the pace:
- De novo peptide generation models create entirely new sequences not found in nature, optimized for specific bacterial targets
- Multi-objective optimization simultaneously maximizes antimicrobial potency while minimizing human cell toxicity and maximizing stability
- Transfer learning models trained on broad antimicrobial data are fine-tuned for specific pathogens, enabling targeted drug design
The limitation is that computational prediction still requires experimental validation. Many computationally promising AMPs fail in the laboratory, and many that succeed in vitro fail in animal models. ML accelerates the discovery pipeline but does not eliminate the need for rigorous biological testing.
The Clinical Reality: Why Aren't AMPs in Every Hospital?
Despite decades of research and thousands of characterized peptides, only a handful of AMPs have reached clinical use. Nisin (a bacteriocin used as a food preservative), gramicidin (a topical antibiotic), polymyxins (last-resort intravenous antibiotics for multidrug-resistant gram-negative infections), and daptomycin (a lipopeptide antibiotic) are among the few.[4]
Several factors explain this gap between laboratory promise and clinical reality:
Proteolytic degradation. AMPs are peptides, and the human body is full of proteases that degrade peptides. Serum half-lives are often measured in minutes. Strategies to address this include D-amino acid substitution (which makes peptides invisible to most human proteases), cyclization, peptidomimetics, and nanoparticle or liposomal delivery systems.[6]
Mammalian cell toxicity. The same properties that allow AMPs to disrupt bacterial membranes, positive charge and amphipathicity, can also damage human cell membranes at higher concentrations. The therapeutic window between bactericidal and cytotoxic concentrations is often narrow. Rational design approaches, like the dual-function FPON peptide that selectively targets gram-negative membranes, are widening this window.[3]
Manufacturing cost. Solid-phase peptide synthesis is expensive at pharmaceutical scale. Recombinant production in bacteria or yeast can reduce costs but introduces challenges with peptide folding, purification, and endotoxin contamination.
Route of administration. Most clinical success has been topical. Systemic delivery faces the combined challenges of proteolytic degradation, renal clearance, and potential immunogenicity. This limits AMPs primarily to skin infections, wound care, and mucosal surfaces, though emerging delivery technologies are expanding the possibilities.
Regulatory pathway. AMPs do not fit neatly into existing regulatory categories. They are biologics (peptides) that act like small-molecule antibiotics. The FDA approval pathway can be uncertain and expensive.
Safety
ModerateMammalian cell toxicity is the biggest hurdle for systemic AMPs
Concern
The same cationic-amphipathic properties that let AMPs lyse bacterial membranes can also damage human cell membranes at higher concentrations. Hemolysis (red blood cell destruction) is the most common dose-limiting toxicity. The therapeutic window — kill bacteria without killing the patient's cells — is often narrow.
What the research says
Engineering progress is widening the window: D-amino acid substitution, stapling, liposomal encapsulation, and dual-trigger designs all help. Topical applications avoid the systemic toxicity issue entirely, which is why most clinical wins so far are topical.
Particularly relevant for: Anyone in early-phase AMP trials, especially systemic administration studies
What to do
Don't equate 'natural origin' with 'safe.' AMPs go through the same toxicity testing as any other drug. At the wrong dose, the difference between killing bacteria and damaging human cells can be narrow.
Hancock & Sahl (2006); Eladl et al. (2025); toxicology reviews of AMP clinical candidates
These are genuine obstacles, not reasons for dismissal. Each has active research programs developing solutions, and the urgency of antimicrobial resistance continues to increase the incentive for regulatory and commercial investment.
The Bottom Line
Antimicrobial peptides represent a fundamentally different approach to fighting bacterial infections than conventional antibiotics. The evidence for their membrane-disrupting mechanisms, broad-spectrum activity, and capacity for synergy with existing drugs is strong and growing. The evidence for their clinical viability is more mixed: real obstacles in stability, toxicity, cost, and delivery have limited therapeutic translation despite decades of research. The most promising near-term applications are AMP-antibiotic combination therapies, topical formulations using advanced delivery systems, and immunomodulatory peptides that boost host defenses rather than replacing antibiotics. Machine learning is accelerating the pace of discovery and optimization. AMPs will not replace antibiotics wholesale, but they are increasingly likely to become part of the antimicrobial arsenal alongside them.
Sources & References
- 1RPEP-01016·Brogden, Kim A (2005). “How Antimicrobial Peptides Kill Bacteria: It's Not Just About Making Holes.” Nature reviews. Microbiology.Study breakdown →PubMed →↩
- 2RPEP-05723·Rima, Mariam et al. (2021). “Antimicrobial Peptides: A Potent Alternative to Antibiotics..” Antibiotics (Basel.Study breakdown →PubMed →↩
- 3RPEP-16223·Tang, Yingqi et al. (2026). “Design and evaluation of dual-function antimicrobial peptides FPON for gram-negative bacteria with membrane disruption and translation inhibition abilities..” mSphere.Study breakdown →PubMed →↩
- 4RPEP-01144·Hancock, Robert E W et al. (2006). “Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies..” Nature biotechnology.Study breakdown →PubMed →↩
- 5RPEP-03618·Cheung, Gordon Y C et al. (2018). “Staph Bacteria Use a Pump to Resist the Body's Antimicrobial Peptides — and This Resistance Drives Real Infections.” The Journal of infectious diseases.Study breakdown →PubMed →↩
- 6RPEP-06225·Javia, Ankit et al. (2022). “Liposomes encapsulating novel antimicrobial peptide Omiganan: Characterization and its pharmacodynamic evaluation in atopic dermatitis and psoriasis mice model..” International journal of pharmaceutics.Study breakdown →PubMed →↩
- 7RPEP-13821·Tongaonkar, Prasad et al. (2025). “Cyclic defensin peptide RTD-1 activates interferon antiviral pathways and blocks SARS-CoV-2 infection in human cells.” Journal of leukocyte biology.Study breakdown →PubMed →↩
- 8RPEP-14721·Al Bouni, Mohamad Anas et al. (2026). “A plant-derived antimicrobial peptide with multiple mechanisms of action exhibiting antibacterial and antibiofilm activities comparable to or superior to polymyxin B..” Current research in microbial sciences.Study breakdown →PubMed →↩
- 9RPEP-11963·Kurbatfinski, Nikola et al. (2025). “Breaking Apart Biofilms Makes MRSA and Other Bacteria Vulnerable to Natural Antimicrobial Peptides.” Microbiology spectrum.Study breakdown →PubMed →↩
- 10RPEP-16012·Roque-Borda, Cesar Augusto et al. (2026). “Synergistic combinations of antimicrobial peptides and conventional antibiotics: A strategy to delay resistance emergence in World Health Organization priority bacteria..” Pharmacological reviews.Study breakdown →PubMed →↩
- 11RPEP-10830·Eladl, Omar (2025). “Biophysical and transcriptomic characterization of LL-37-derived antimicrobial peptide targeting multidrug-resistant Escherichia coli and ESKAPE pathogens..” Scientific reports.Study breakdown →PubMed →↩
- 12RPEP-09464·Wan, Fangping et al. (2024). “How AI and Machine Learning Are Accelerating the Discovery of Bacteria-Killing Peptides.” Nature reviews bioengineering.Study breakdown →PubMed →↩