Anticancer Peptides: How They Kill Tumor Cells
Anticancer Peptides
1,000+ clinical trials
Over 1,000 peptide-based cancer trials are registered in the NIH database, with more than 20 anticancer peptides approved by the FDA and EMA.
Xie et al., Open Biology, 2020
Xie et al., Open Biology, 2020
If you only read one thing
Anticancer peptides are short chains of amino acids engineered to kill cancer cells while leaving healthy cells alone. They work because cancer cells have a different 'fingerprint' on their surface — more negative charge, weaker membrane, and exposed markers that healthy cells hide. Unlike traditional chemo, which hits every fast-dividing cell (including hair and gut lining), peptides aim at what makes cancer chemically different. Over 20 are already FDA-approved, and new classes like 'stapled peptides' are reaching parts of cancer cells that older drugs couldn't touch. The main limits right now are cost, serum stability, and getting enough of the drug into deep solid tumors.
Cancer cells are not just fast-dividing normal cells. Their membranes carry a molecular signature that sets them apart: an outer surface studded with negatively charged phospholipids, higher concentrations of certain glycoproteins, and altered cholesterol content. Anticancer peptides exploit these differences. These short sequences of 10 to 60 amino acids can distinguish malignant cells from healthy tissue and destroy them through mechanisms that conventional chemotherapy cannot easily replicate. Over 1,000 peptide-based cancer clinical trials are now registered, and more than 20 anticancer peptides have received FDA or EMA approval.[1] This article maps the full landscape of how these peptides work, from membrane-lytic killing to intracellular target engagement to immune system activation.
Key Takeaways
- Cancer cells have a different surface chemistry than healthy cells — more negative charge, weaker membrane, exposed markers healthy cells hide.
- Anticancer peptides exploit that difference. They're short amino acid chains engineered to stick to cancer cells while leaving healthy ones alone.
- This is what makes them different from chemo. Chemo kills every fast-dividing cell, including hair and gut lining. Peptides aim at a chemical signature.
- Over 20 anticancer peptides are already FDA-approved, and more than 1,000 peptide cancer trials are running worldwide right now.
- In one mouse experiment, a membrane-attacking peptide wiped out melanoma tumors in every treated animal — and trained the immune system to attack future tumors.
- Another type, called "stapled peptides," reaches deep inside cancer cells to reactivate the tumor-suppressor gene p53 — something no traditional drug can do.
- Cancer has a harder time evolving resistance to peptide drugs than to chemo, because membrane-attack mechanisms are much harder for tumors to work around.
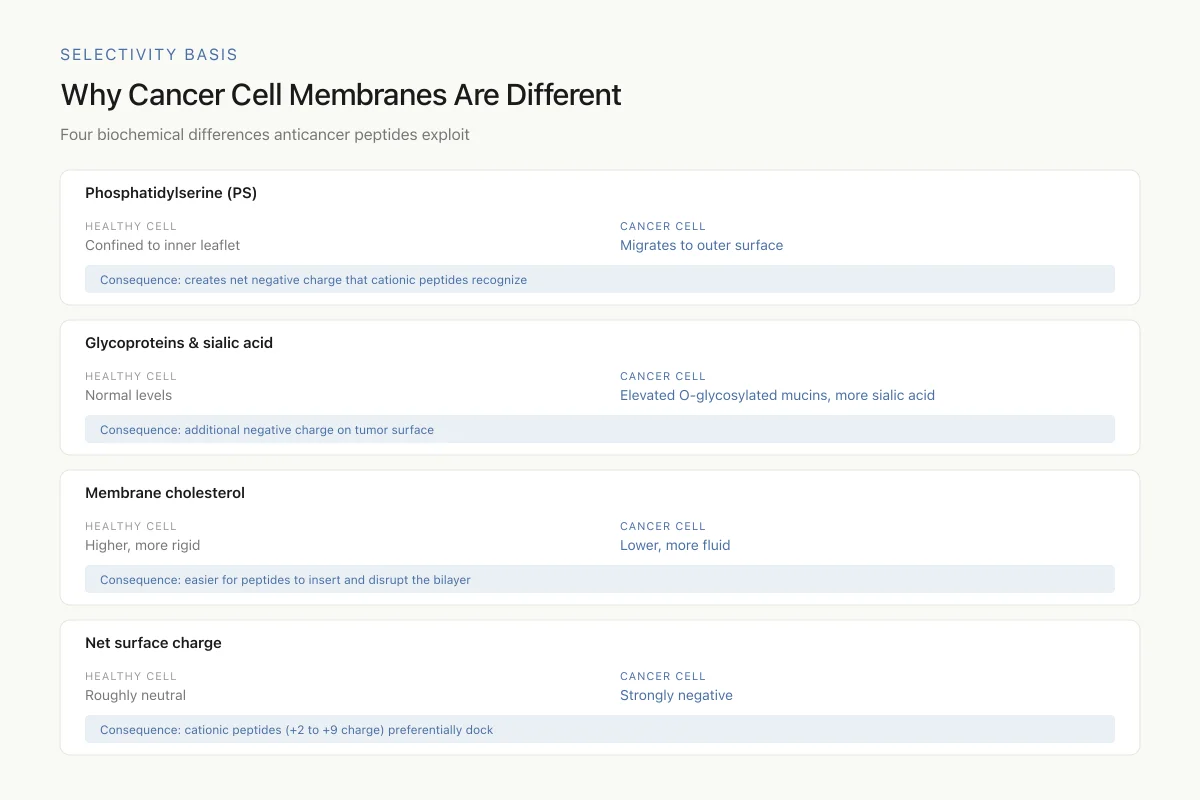
Why cancer cell membranes are different
The selectivity of anticancer peptides begins with a fundamental biochemical asymmetry. Healthy mammalian cells maintain an asymmetric lipid distribution across their plasma membrane: negatively charged phosphatidylserine (PS) is confined almost exclusively to the inner leaflet. Cancer cells lose this asymmetry. PS migrates to the outer membrane surface, creating a net negative charge that cationic peptides recognize and bind.[9]
This is not a subtle difference. Riedl et al. (2015) demonstrated that human lactoferricin-derived peptides specifically targeted PS-enriched cancer cell membranes of melanoma, glioblastoma, and rhabdomyosarcoma lines while leaving non-cancerous cells intact. When they engineered peptides with loop structures that enhanced PS binding, cancer cell killing improved with no increase in toxicity to healthy fibroblasts.[9]
Beyond PS exposure, cancer cells also display increased numbers of O-glycosylated mucins, higher levels of sialic acid-bearing glycoproteins, and elevated heparan sulfate proteoglycans on their surfaces. These molecules contribute additional negative charge. At the same time, cancer cell membranes typically contain less cholesterol than healthy cells, making them more fluid and susceptible to peptide insertion and disruption.[1]
Selectivity Basis
Why Cancer Cell Membranes Are Different
Four biochemical differences between tumor and healthy membranes that anticancer peptides exploit
Normal chemotherapy hits every fast-dividing cell — cancer, hair follicles, gut lining. Anticancer peptides aim at what makes cancer *chemically* different, not what makes it divide fast. That's a fundamentally different kind of selectivity.
Source: Xie et al. (2020), Open Biology; Riedl et al. (2015)
 View as image
View as imageThe net result: a cancer cell membrane is both more negatively charged and more physically vulnerable than a normal cell membrane. Cationic anticancer peptides, typically carrying a net charge of +2 to +9, preferentially accumulate on these surfaces through electrostatic attraction before executing their lethal mechanisms.
How amphipathic peptides destroy cancer cell membranes
The largest and most studied class of anticancer peptides kills cancer cells by physically tearing apart their membranes. These amphipathic peptides carry both positively charged and hydrophobic regions arranged along an alpha-helical backbone. After electrostatic docking to the cancer cell surface, the hydrophobic face inserts into the lipid bilayer.
Three primary models describe what happens next. In the barrel-stave model, peptides insert perpendicularly through the membrane to form transmembrane pores lined by peptide molecules. In the carpet model, peptides accumulate parallel to the membrane surface until reaching a threshold concentration that causes catastrophic membrane dissolution. In the toroidal pore model, peptides induce the lipid molecules themselves to curve inward, forming pores lined by both peptide and lipid headgroups.[2]
Bui et al. (2025) explored these mechanisms using Mastoparan AF derivatives, cationic amphipathic peptides from wasp venom. Through systematic amino acid modifications, they increased the selectivity of these peptides for cancer cell membranes over erythrocytes. Circular dichroism spectroscopy confirmed that modifications preserving the alpha-helical conformation maintained anticancer potency, while those disrupting helicity reduced membrane-lytic activity. They also combined modified Mastoparan derivatives with the oncolytic peptide LTX-315 and observed synergistic killing of cancer cells.[5]
A particularly inventive approach to the selectivity problem came from Ning et al. (2026), who engineered Charge-Alternating Spherical Membrane-Lytic Peptide (CAS-MLP) bottlebrushes. These nanostructures masked the membrane-lytic peptides behind a pH-sensitive shield. At the slightly acidic pH found in tumor microenvironments (pH 6.5-6.8), the shield unmasked, exposing the active peptide. The redox-responsive backbone then degraded in the presence of intracellular glutathione, releasing individual membrane-lytic peptide units. This dual-gating system achieved selective tumor lysis while dramatically reducing the hemolysis that limits conventional membrane-active peptides.[14]
Melittin, a 26-amino acid peptide from bee venom, exemplifies both the promise and the challenge. Yue et al. (2026) used molecular dynamics simulations to study how melittin interacts with cancer cell membranes enriched in sialic acid. They found that sialic acid on cancer cell surfaces provided initial binding sites for the cationic peptide, increasing local peptide concentration and accelerating pore formation. When combined with cold atmospheric plasma treatment, the synergistic membrane damage exceeded what either approach achieved alone.[15]
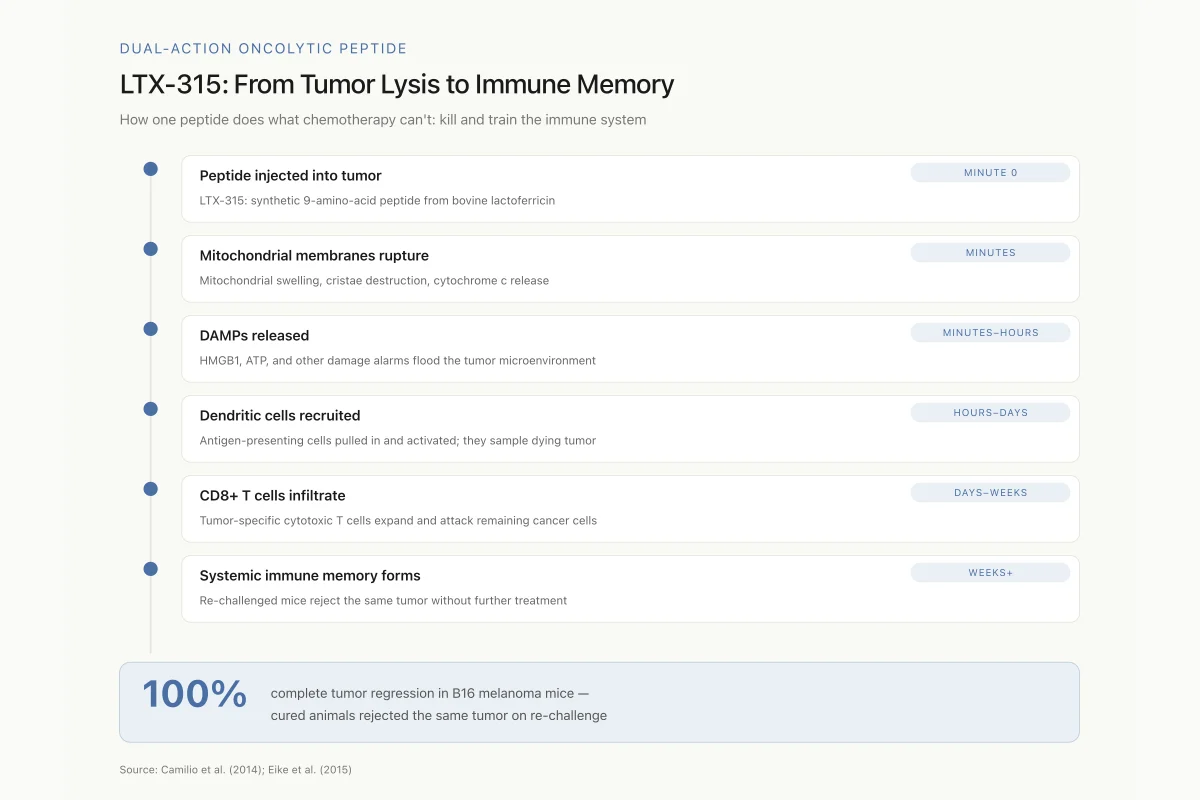
LTX-315: an oncolytic peptide that triggers anti-tumor immunity
Not all membrane-disrupting peptides simply lyse cancer cells and stop there. LTX-315, a synthetic nonapeptide derived from structure-activity studies on bovine lactoferricin, represents a class of oncolytic peptides that kill tumor cells and simultaneously activate the immune system against surviving cancer.
Eike et al. (2015) showed that LTX-315 killed human melanoma cells not by classical apoptosis but by inducing mitochondrial membrane disruption. The peptide caused massive mitochondrial swelling, cristae destruction, and release of cytochrome c. This mitochondria-targeted cell death triggered the release of damage-associated molecular patterns (DAMPs), including HMGB1 and ATP, molecular alarm signals that recruit and activate dendritic cells.[6]
Dual-Action Oncolytic Peptide
LTX-315: From Tumor Lysis to Immune Memory
How one peptide does what chemotherapy can't: kill the tumor and train the immune system to finish the job
This is why oncolytic peptides are paired with checkpoint immunotherapy in trials. The peptide does the initial debulking and releases tumor antigens in a way that feels like an infection to the immune system. Checkpoint inhibitors then keep the T cell response running.
Source: Camilio et al. (2014); Eike et al. (2015)
 View as image
View as imageThe immunological consequences proved striking. Camilio et al. (2014) treated B16 melanoma-bearing mice with intratumoral LTX-315 injections and achieved complete tumor regression in treated animals. More remarkably, when the researchers re-challenged cured mice with the same tumor cells, the animals rejected the tumors without additional treatment. Analysis revealed that LTX-315 treatment had generated a systemic, long-lasting anti-tumor immune response dominated by CD8+ cytotoxic T cells infiltrating the tumor site.[7]
This dual action, direct tumor killing plus immune activation, positions oncolytic peptides as potential partners for checkpoint immunotherapy. The peptide does the initial tumor destruction and releases tumor antigens in an immunogenic context; the immune system does the long-term surveillance.
Pro-apoptotic peptides: targeting the mitochondrial death pathway
While membrane-lytic peptides attack the plasma membrane from outside, pro-apoptotic peptides penetrate the cell and trigger its internal self-destruction program. The mitochondrial pathway of apoptosis is a particularly attractive target because cancer cells often evade death by overexpressing anti-apoptotic proteins like BCL-2, BCL-XL, and MCL-1.
The KLA peptide, with the sequence (KLAKLAK)₂, is among the most studied pro-apoptotic anticancer peptides. This 14-residue amphipathic sequence has minimal effect on the plasma membrane at therapeutic concentrations but rapidly disrupts mitochondrial membranes once inside the cell, collapsing the mitochondrial membrane potential and triggering cytochrome c release. Cho et al. (2026) reviewed decades of KLA research and found that the peptide's main limitation, poor cellular uptake, has been addressed through conjugation with cell-penetrating peptides, tumor-homing sequences, and nanoparticle delivery systems. These delivery strategies improved tumor targeting while maintaining the peptide's mitochondrial specificity.[8]
The BH3 domain represents another pro-apoptotic strategy. BCL-2 family proteins regulate mitochondrial apoptosis through protein-protein interactions mediated by BH3 domains, short alpha-helical segments that fit into hydrophobic grooves on anti-apoptotic proteins. Walensky et al. (2004) introduced hydrocarbon stapling to stabilize BH3 peptides, creating SAHBs (Stabilized Alpha-Helix of BCL-2 domains). These stapled peptides bound BCL-2 family proteins with enhanced affinity, resisted protease degradation, penetrated cell membranes, and activated apoptosis in leukemia cells both in vitro and in xenograft models.[3]
This work established a principle: locking peptides into their bioactive alpha-helical conformation through chemical stapling creates drug-like molecules from sequences that would otherwise be too unstable and impermeable for therapeutic use.
Stapled peptides: engineering stability into anticancer peptides
Stapled peptides address three fundamental problems that limit conventional peptides as drugs: rapid protease degradation, poor membrane permeability, and loss of the alpha-helical structure required for target binding. The hydrocarbon staple, a covalent bridge between amino acid side chains spanning one or two helical turns, constrains the peptide backbone and simultaneously creates a hydrophobic surface that facilitates cell entry.
The clinical validation of this technology came with sulanemadlin (ALRN-6924). Guerlavais et al. (2023) described its development as the first cell-permeating, stabilized alpha-helical peptide to enter clinical trials. Sulanemadlin mimics the N-terminal transactivation domain of p53 and binds with high affinity to both MDM2 and MDMX, the two endogenous inhibitors of the p53 tumor suppressor. In tumors that retain wild-type p53 but overexpress MDM2 or MDMX, the peptide releases p53 from inhibition, reactivating the apoptotic program.[4]
Choudhury et al. (2025) investigated how different hydrocarbon cross-linker lengths and positions affect the structure and binding of stapled p53 peptides to MDM2. Using molecular dynamics simulations, they found that the staple position relative to the binding interface critically determined both helical stability and binding affinity. Staples placed too close to the binding residues could sterically interfere with MDM2 engagement, while those placed too far away failed to stabilize the critical binding helix. The optimal positioning balanced structural rigidity with conformational freedom at the binding interface.[12]
Beyond p53-MDM2, stapled peptides are being developed against other intracellular cancer targets, including the p53-MDM2/MDMX axis, BCL-2 family interactions, and RAS-RAF signaling. The technology platform is flexible: the same stapling chemistry can be applied to any alpha-helical peptide interface, making it broadly applicable across intracellular protein-protein interactions in oncology.
Peptides targeting p53 pathway reactivation
The p53 tumor suppressor is mutated or functionally inactivated in the majority of human cancers. In the approximately 50% of cancers that retain wild-type p53, the protein is often suppressed by overexpressed MDM2 or MDMX, which bind p53 and mark it for proteasomal degradation. Peptides that block this interaction can reactivate p53 signaling and restore the cancer cell's ability to undergo apoptosis.
Philippe et al. (2021) developed "Angler Peptides," macrocyclic conjugates designed to inhibit both p53:MDM2 and p53:MDMX interactions. By conjugating high-affinity p53-mimicking peptides with cell-penetrating peptide sequences, they achieved cytosolic delivery and demonstrated apoptosis activation in cancer cells. The macrocyclic structure provided both protease resistance and conformational constraint, improving binding affinity over linear peptide counterparts.[11]
Kang et al. (2025) took a different approach, targeting the FOXO4-p53 interaction instead of MDM2-p53. In senescent cancer cells, FOXO4 binds and sequesters p53 in the nucleus, preventing it from triggering apoptosis. Their optimized peptide, CPP-CAND, disrupted this interaction and induced apoptosis selectively in senescent cancer cells while sparing non-senescent cells. This selectivity for cellular senescence represents a fundamentally different targeting strategy: rather than distinguishing cancer from normal tissue by membrane composition, the peptide distinguishes by cellular state.[13]
Natural peptides with anticancer activity
Many anticancer peptide candidates derive from natural host defense peptides found across species. These molecules evolved to kill pathogens through membrane disruption, and their selectivity for negatively charged membranes translates into preferential activity against cancer cells.
Lactoferricin, derived from bovine lactoferrin, has been extensively studied for anticancer activity. Grissenberger et al. (2020) designed optimized human lactoferricin derivatives and tested them against malignant melanoma in both conventional 2D cultures and 3D tumor spheroid models. The peptides induced apoptosis in melanoma cells and specifically targeted lipid-enriched membrane domains. Testing in 3D spheroids, which better mimic the tumor microenvironment, confirmed activity under more physiologically relevant conditions.[10]
Cecropin-magainin hybrid peptides combine structural elements from insect (cecropin A) and amphibian (magainin 2) antimicrobial peptides. These hybrids consistently show antitumor activity across multiple cancer cell lines while maintaining selectivity over healthy cells. The structure-activity relationship is well characterized: net positive charge drives membrane selectivity, helicity determines membrane insertion depth, and hydrophobicity modulates the balance between anticancer potency and hemolytic toxicity.[2]
Magainin 2, originally isolated from the skin of the African clawed frog Xenopus laevis, was among the earliest antimicrobial peptides shown to kill cancer cells. Its IC50 against lung cancer A549 cells is approximately 110 micrograms per milliliter, higher than the concentrations achievable in vivo without modification. This illustrates a recurring theme: natural anticancer peptides often possess the right selectivity mechanism but lack the potency or stability for direct clinical use. The value of studying them lies in identifying the structural principles, charge distribution, amphipathic helix geometry, and membrane interaction dynamics, that can be optimized in synthetic derivatives.[1]
Alpha-defensins, the small cationic peptides released by neutrophils during innate immune responses, also demonstrate anticancer properties. Their beta-sheet structures and disulfide-stabilized folds give them membrane-penetrating abilities distinct from the alpha-helical ACPs, expanding the structural repertoire of peptide-based cancer therapeutics.
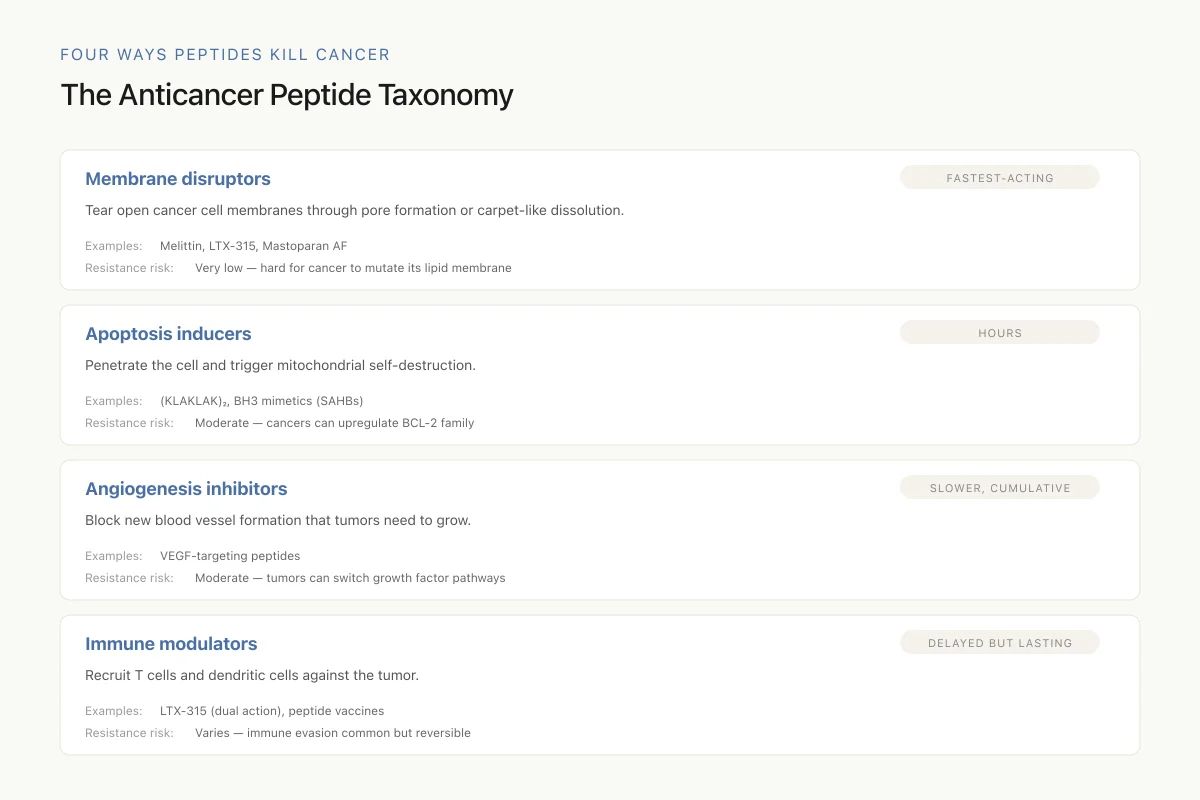
Classification of anticancer peptides by mechanism
Xie et al. (2020) proposed a systematic classification of ACPs into four functional categories:[1]
Four Ways Peptides Kill Cancer
The Anticancer Peptide Taxonomy
Many real-world anticancer peptides don't stay in one category. LTX-315, for example, is both a membrane disruptor and an immune modulator — it kills the tumor directly and recruits T cells for the long-term cleanup. This multi-modal behavior is part of why peptides are harder for cancer to outmaneuver.
Source: Xie et al. (2020); Gaspar et al. (2013), Biochim Biophys Acta
 View as image
View as imageMembrane disruptors physically breach the cancer cell membrane through pore formation or carpet-like dissolution. This is the fastest-acting mechanism and the most resistant to drug resistance development because it targets the lipid membrane itself rather than a specific protein.
Apoptosis inducers trigger programmed cell death through mitochondrial membrane disruption, caspase cascade activation, or release of pro-apoptotic factors. These peptides must penetrate the plasma membrane to reach intracellular targets.
Angiogenesis inhibitors block the formation of new blood vessels that tumors require for sustained growth. Some peptides target VEGF receptors or downstream signaling; others interfere with endothelial cell migration.
Immune modulators activate anti-tumor immune responses by stimulating dendritic cell maturation, enhancing T cell cytotoxicity, or releasing tumor antigens in immunogenic contexts. LTX-315 falls into both the membrane disruptor and immune modulator categories.
Gaspar et al. (2013) noted that many anticancer peptides originally identified as antimicrobial peptides exhibit multiple mechanisms simultaneously. A single peptide might disrupt the plasma membrane at high concentrations, trigger mitochondrial apoptosis at lower concentrations, and activate immune cells through DAMP release. This multi-modal activity is both an advantage, it reduces resistance risk, and a challenge for rational drug design.[2]
Current limitations and open questions
Anticancer peptides face several obstacles between the laboratory and the clinic. Serum stability remains the most universal challenge: unmodified peptides are degraded by proteases within minutes of entering the bloodstream. Strategies to address this include D-amino acid substitution, cyclization, hydrocarbon stapling, and PEGylation, each with trade-offs in cost, activity, and immunogenicity.
Safety
ModerateHemolysis is the most common dose-limiting toxicity
Concern
Membrane-lytic anticancer peptides are cationic — positively charged peptides that target negatively charged membranes. Cancer cells are the preferred target, but at high enough concentrations, red blood cells (also slightly anionic) get hit too. Hemolysis is the dose-ceiling for most peptides in this class.
What the research says
Engineering solutions are working: pH-gated bottlebrush peptides (CAS-MLP), D-amino acid substitution, and stapling have all widened the therapeutic window. But the concept of a 'natural, gentle' peptide therapy is wrong — these drugs have real dose limits enforced by the kidneys and the blood.
Particularly relevant for: Patients in anticancer peptide trials, especially those with baseline anemia or kidney dysfunction
What to do
Trials monitor hematocrit, haptoglobin, and free hemoglobin closely. Don't assume 'peptide = natural = no red blood cell risk.'
Gaspar et al. (2013); Bui et al. (2025); Ning et al. (2026)
Selectivity is imperfect. While the electrostatic basis of cancer cell targeting is well established, ACPs are not binary switches. At high enough concentrations, most cationic peptides will damage healthy cell membranes. The therapeutic window, the concentration range that kills cancer cells without significant toxicity to normal tissue, varies widely across peptides and cancer types. Hemolysis of red blood cells is the most common dose-limiting toxicity.
Tumor penetration presents another barrier, particularly for solid tumors. Peptides that work well on isolated cancer cells or small tumor spheroids may fail against large, poorly vascularized tumors with dense stromal tissue. Delivery technologies including nanoparticle encapsulation, peptide-drug conjugates, and tumor-homing sequences are being developed to address tissue-level distribution.
Manufacturing cost exceeds that of small-molecule drugs, though advances in recombinant production and solid-phase synthesis have reduced prices considerably. The relatively short sequences of most ACPs (10-60 amino acids) make them more accessible than larger biologic drugs like antibodies.
The evidence base, while growing rapidly, still relies heavily on in vitro studies and mouse models. Translation to human clinical outcomes has been slower than the preclinical data would suggest. The gap between IC50 values measured in cell culture and effective concentrations achievable in human tumors remains one of the largest uncertainties in the field. Representative IC50 values from cell culture span a wide range: aurein 1.2 showed activity at 2 micromolar against glioblastoma T98G cells, while magainin II required 110 micrograms per milliliter against lung cancer A549 cells. Modified peptides have closed this gap; for example, the S25K modification reduced IC50 from 13.2 micromolar to 1.4 micromolar in cervical cancer cells.[1] Whether these optimized in vitro potencies translate to effective tumor concentrations in patients remains an open question for each candidate.
Combination strategies may offer a path forward. Pairing anticancer peptides with conventional chemotherapeutics, checkpoint inhibitors, or other peptides (as Bui et al. demonstrated with Mastoparan AF and LTX-315 combinations) can achieve synergistic killing at lower individual peptide concentrations, potentially widening the therapeutic window.[5]
The Bottom Line
Anticancer peptides exploit fundamental differences between cancer and normal cell membranes to achieve selective tumor killing. The evidence spans membrane-lytic peptides that physically destroy cancer cells, pro-apoptotic sequences that trigger mitochondrial death pathways, stapled peptides that reach intracellular targets like p53-MDM2, and oncolytic peptides that combine direct killing with immune activation. Over 20 ACPs have reached clinical approval, and sulanemadlin demonstrated that stapled peptides can enter clinical development as a viable drug class. The field's main unresolved challenges are serum stability, achieving sufficient therapeutic windows, and translating preclinical potency into clinical efficacy in solid tumors.
Sources & References
- 1RPEP-05206·Xie, Mingfeng et al. (2020). “Anti-cancer peptides: classification, mechanism of action, reconstruction and modification..” Open biology.Study breakdown →PubMed →↩
- 2RPEP-02177·Gaspar, Diana et al. (2013). “From antimicrobial to anticancer peptides. A review..” Frontiers in microbiology.Study breakdown →PubMed →↩
- 3RPEP-00992·Walensky, Loren D et al. (2004). “Stapled Peptide Helix Activates Cancer Cell Death In Vivo: A Breakthrough in Peptide Drug Design.” Science (New York.Study breakdown →PubMed →↩
- 4RPEP-06925·Guerlavais, Vincent et al. (2023). “Discovery of Sulanemadlin (ALRN-6924), the First Cell-Permeating, Stabilized α-Helical Peptide in Clinical Development..” Journal of medicinal chemistry.Study breakdown →PubMed →↩
- 5RPEP-10244·Bui Thi Phuong, Hai et al. (2025). “Developing Potent and Selective Anticancer Therapy through Chemical Approaches and the Combination of Cationic Amphipathic Oncolytic Peptides..” Journal of medicinal chemistry.Study breakdown →PubMed →↩
- 6RPEP-02619·Eike, Liv-Marie et al. (2015). “The oncolytic peptide LTX-315 induces cell death and DAMP release by mitochondria distortion in human melanoma cells..” Oncotarget.Study breakdown →PubMed →↩
- 7RPEP-02345·Camilio, Ketil André et al. (2014). “Complete regression and systemic protective immune responses obtained in B16 melanomas after treatment with LTX-315..” Cancer immunology.Study breakdown →PubMed →↩
- 8RPEP-15035·Cho, Yunmi et al. (2026). “New Nanotech Delivery Systems for the Cancer-Fighting KLA Peptide.” Biomolecules.Study breakdown →PubMed →↩
- 9RPEP-02778·Riedl, Sabrina et al. (2015). “Milk-Derived Peptides Selectively Kill Cancer Cells by Targeting a Cancer-Specific Membrane Marker.” Biochimica et biophysica acta.Study breakdown →PubMed →↩
- 10RPEP-04828·Grissenberger, Sarah et al. (2020). “Modified Lactoferricin Peptides Selectively Kill Melanoma While Sparing Normal Cells.” Biochimica et biophysica acta. Biomembranes.Study breakdown →PubMed →↩
- 11RPEP-05685·Philippe, Grégoire J-B et al. (2021). “Angler Peptides: Macrocyclic Conjugates Inhibit p53:MDM2/X Interactions and Activate Apoptosis in Cancer Cells..” ACS chemical biology.Study breakdown →PubMed →↩
- 12RPEP-10480·Choudhury, Asha Rani et al. (2025). “Exploring the Effect of Hydrocarbon Cross-Linkers on the Structure and Binding of Stapled p53 Peptides..” Proteins.Study breakdown →PubMed →↩
- 13RPEP-11705·Kang, Donghoon et al. (2025). “Designed Peptide Selectively Kills Senescent Cancer Cells That Survive Chemotherapy.” Journal of medicinal chemistry.Study breakdown →PubMed →↩
- 14RPEP-15800·Ning, Lubin et al. (2026). “A Smart Nanoparticle That Keeps Cancer-Killing Peptides Dormant Until They Reach the Tumor.” Journal of controlled release : official journal of the Controlled Release Society.Study breakdown →PubMed →↩
- 15RPEP-16521·Yue, Yi et al. (2026). “Synergistic mechanism of plasma and melittin-induced membrane perforation in sialic acid-targeted cancer cells..” The Journal of chemical physics.Study breakdown →PubMed →↩