Telomeres and Aging: The Biological Clock in Your DNA
Telomeres & Aging
TTAGGG repeat
The six-nucleotide DNA sequence repeated thousands of times at the end of every human chromosome. Each cell division shortens these repeats until the cell can no longer divide.
Blackburn et al., Nobel Prize in Physiology or Medicine, 2009
Blackburn et al., Nobel Prize in Physiology or Medicine, 2009
If you only read one thing
Your chromosomes have protective caps called telomeres that get shorter every time a cell divides. When they get too short, the cell stops working properly and starts pumping out inflammatory junk that ages your whole body. A tiny peptide called epitalon can reactivate the enzyme that rebuilds these caps — and it extended lifespan by up to 31% in animals. But here's the reality check: no large human trial has ever been done. The cell and animal science is real, but 'proven anti-aging treatment' is a stretch when the research mostly comes from one lab and has never been tested at scale in people.
Every time a human cell divides, it loses 50 to 200 base pairs from the ends of its chromosomes. These ends, called telomeres, are repetitive DNA sequences (TTAGGG in vertebrates) that protect coding genes from degradation. When telomeres become critically short, cells enter a state called replicative senescence: they stop dividing, change their secretory profile, and contribute to tissue aging. This is the Hayflick limit, discovered in 1961, and telomere shortening is its molecular mechanism.
The enzyme telomerase can rebuild telomeres by adding TTAGGG repeats back to chromosome ends. It is active in stem cells and germ cells but silenced in most adult somatic cells, which is why those cells age. The 2009 Nobel Prize in Physiology or Medicine was awarded to Elizabeth Blackburn, Carol Greider, and Jack Szostak for discovering telomerase and establishing the connection between telomere maintenance and cellular aging.
The peptide connection enters here: the tetrapeptide epitalon (Ala-Glu-Asp-Gly) activates telomerase in human somatic cells and extends telomere length beyond normal replicative limits.[1] Whether this translates to measurable anti-aging effects in humans is the question this article examines.
For details on the specific peptide approaches to telomere maintenance, see Can Peptides Activate Telomerase? The Science of Telomere Maintenance. For the evidence on what telomere length measurements actually indicate, see Telomere Length as an Aging Biomarker: What It Tells Us and What It Doesn't.
Key Takeaways
- Your chromosomes have protective caps called telomeres — like the plastic tips on a shoelace.
- Every time a cell divides, it loses a tiny bit of that cap and eventually runs out.
- When telomeres get too short, cells turn into "zombies" that leak inflammation into the rest of your body.
- A peptide called epitalon can reactivate the enzyme that rebuilds those caps — at least in a petri dish.
- In fruit flies it extended lifespan by 31%; in mice, by about 12%.
- No large human trial of epitalon has ever been done — almost all evidence comes from one Russian lab.
- Reactivating telomerase walks a tightrope: about 85% of cancers use the same enzyme to grow forever.
Telomere Structure: The Protective Cap
Biological Clock
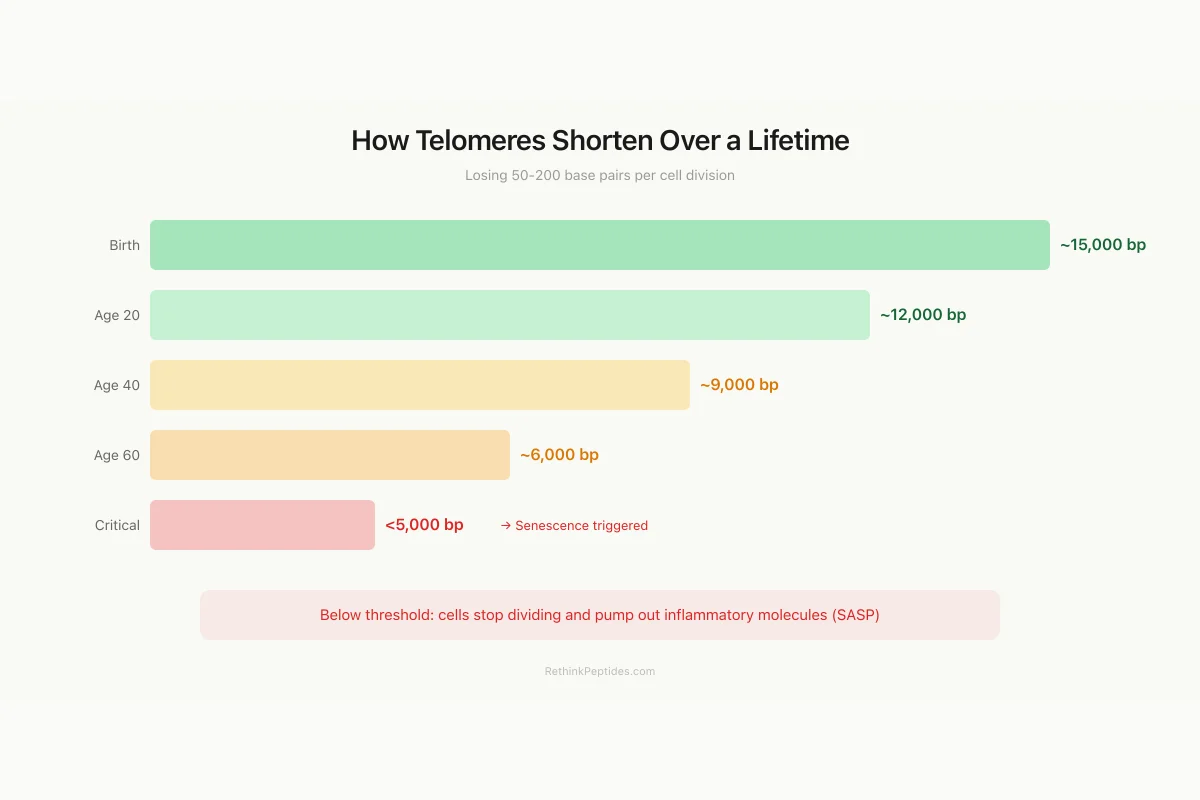
How Telomeres Shorten Over a Lifetime
Losing 50-200 base pairs per cell division, from birth to senescence
Below ~5,000 bp, cells trigger a p53/p21 damage response and stop dividing (senescence). Senescent cells then pump out inflammatory molecules that age surrounding tissue — the senescence-associated secretory phenotype (SASP).
Source: Blackburn et al. (2009 Nobel); estimated leukocyte telomere ranges
 View as image
View as imageTelomeres are not simply passive buffer zones of disposable DNA. They are dynamic nucleoprotein structures with a specific architecture that protects chromosome integrity.
Each human telomere consists of 5,000 to 15,000 tandem repeats of the hexanucleotide TTAGGG. The double-stranded telomeric DNA terminates in a single-stranded 3' overhang of 50 to 300 nucleotides, which folds back and invades the double-stranded region to form a T-loop structure. This loop physically hides the chromosome end from the DNA damage machinery that would otherwise recognize it as a broken DNA molecule and attempt to repair it through non-homologous end joining, fusing chromosomes together.
The T-loop is stabilized by the shelterin complex, a group of six proteins (TRF1, TRF2, TIN2, TPP1, POT1, and RAP1) that bind telomeric DNA and regulate telomerase access. TRF2 prevents activation of the ATM kinase damage response. POT1 binds the single-stranded overhang and prevents ATR kinase activation. When shelterin function is compromised, even telomeres of normal length trigger a DNA damage response, demonstrating that telomere function depends on protein-DNA structure, not just sequence length.
Understanding telomere structure is essential because interventions aimed at extending telomere length, whether through peptides, small molecules, or lifestyle changes, must work within this structural context. Simply adding TTAGGG repeats is insufficient if shelterin cannot properly cap the new sequences. The quality of telomere extension matters as much as the quantity.
The End-Replication Problem
The fundamental reason telomeres shorten is a consequence of how DNA polymerase works. DNA replication requires an RNA primer to initiate synthesis. On the lagging strand, the terminal RNA primer at the chromosome end cannot be replaced with DNA because there is no upstream Okazaki fragment to extend. The result: each round of DNA replication fails to copy the very end of the chromosome, losing approximately 50-200 base pairs per division.
This is the end-replication problem, first articulated by James Watson in 1972 and independently by Alexei Olovnikov. It means that normal somatic cells have a finite replicative capacity. After 40-60 divisions (the Hayflick limit), telomeres become critically short (typically below 4,000-5,000 base pairs), triggering a p53/p21-mediated cell cycle arrest. The cell enters replicative senescence: it remains metabolically active but stops dividing.
Senescent cells are not inert. They secrete a characteristic mix of inflammatory cytokines, matrix metalloproteinases, and growth factors collectively called the senescence-associated secretory phenotype (SASP). SASP drives chronic low-grade inflammation (inflammaging), impairs tissue repair, and contributes to the pathology of age-related diseases including atherosclerosis, osteoarthritis, and neurodegeneration.
Telomerase: The Enzyme That Reverses the Clock
Telomerase is a ribonucleoprotein enzyme composed of a catalytic subunit (TERT, telomerase reverse transcriptase) and an RNA template component (TERC). TERC contains a short sequence complementary to the telomere repeat, and TERT uses it as a template to synthesize new TTAGGG repeats at the chromosome end.
In humans, telomerase is highly active in embryonic stem cells, germ cells, and activated lymphocytes, maintaining their telomere length through continuous division. It is repressed in most adult somatic cells through epigenetic silencing of the TERT promoter. This repression is both a tumor-suppressive mechanism (unlimited replication is a hallmark of cancer) and the molecular basis of replicative aging.
The therapeutic dilemma is that reactivating telomerase in somatic cells could extend their lifespan but might also promote cancer. Approximately 85-90% of human cancers reactivate telomerase as part of their immortalization strategy. The remaining 10-15% use an alternative lengthening of telomeres (ALT) mechanism based on homologous recombination. Any strategy to extend telomeres for anti-aging purposes must contend with this cancer risk.
Epitalon: The Telomerase-Activating Peptide
Epitalon (also spelled epithalon) is a synthetic tetrapeptide (Ala-Glu-Asp-Gly) derived from epithalamin, a peptide extract of the pineal gland. It was developed by Vladimir Khavinson and colleagues at the Saint Petersburg Institute of Bioregulation and Gerontology as part of a broader research program on peptide bioregulators.[2]
The Landmark 2003 Study
Khavinson et al. (2003) published the foundational paper demonstrating that epitalon activates telomerase in human somatic cells.[1] In human fetal lung fibroblasts, epitalon treatment induced telomerase activity, increased telomere length, and allowed cells to exceed the normal Hayflick limit by 10 additional population doublings. Treated cells maintained a youthful morphology and normal karyotype, showing no signs of malignant transformation.
The same group showed that epitalon treatment increased telomere length in blood cells of elderly patients (ages 60-80), with the effect reaching statistical significance in both the 60-65 and 75-80 age groups. The mechanism appeared to involve transcriptional activation of the TERT gene through peptide-DNA interactions.
The 2026 Confirmation
Sanchez et al. (2026) provided modern confirmation and mechanistic refinement of epitalon's effects on telomere length.[3] Published in Biochemical and Biophysical Research Communications, the study demonstrated that epitalon increases telomere length in human cell lines through two distinct pathways: telomerase upregulation (TERT expression) and activation of the ALT pathway. This dual mechanism was unexpected: most telomere-extending interventions work through one pathway or the other, not both.
The study also characterized epitalon as a multi-pathway geroprotector acting on five hallmarks of aging: telomere maintenance, epigenetic regulation, oxidative stress resilience, immune recalibration, and circadian rhythm restoration.
Evidence Summary
What We Know About Epitalon's Telomere Effects
Epitalon activated telomerase in human fibroblasts, extended lifespan by 10 extra doublings beyond Hayflick limit
Confirmed telomere extension via both telomerase upregulation AND alternative lengthening (ALT) — dual mechanism
Epithalamin increased mean lifespan: +31% in fruit flies, +12.3% in mice, +24% in female rats
Epitalon reduced spontaneous tumor incidence ~30% in female mice and extended maximum lifespan
Increased telomere length in blood cells of elderly patients (ages 60-80), statistically significant
Does not exist. No placebo-controlled human trial for anti-aging endpoints.
Source: Khavinson (2003); Sanchez (2026); Anisimov (1998, 2003)
 View as image
View as imageLifespan Studies
Anisimov et al. (1998) tested epithalamin (the pineal extract from which epitalon was derived) in fruit flies, mice, and rats.[4] Mean lifespan increased by 31% in fruit flies, 12.3% in CBA mice, and 24% in female rats. Spontaneous tumor incidence decreased in treated animals. A follow-up study by the same group (2003) showed that epitalon itself (the synthetic tetrapeptide) reduced spontaneous tumor incidence in female mice by approximately 30% and extended maximum lifespan.[5]
Ivko et al. (2025) published a comprehensive review of epitalon's properties, consolidating data from over two decades of research.[6] The review categorized the evidence as strong for in vitro telomerase activation and moderate for in vivo anti-aging effects, while noting that large-scale, randomized, placebo-controlled human trials have not been conducted.
Khavinson's broader research program on peptide bioregulators, including epitalon, is documented in his 2002 monograph "Peptides and Ageing," which proposed that short peptides (2-4 amino acids) can regulate gene expression by binding directly to DNA and modifying transcription of age-related genes.[2] This mechanism remains controversial but has been supported by subsequent molecular modeling and in vitro binding studies.
For epitalon's connection to the pineal gland and melatonin production, see Epithalon and Melatonin: The Pineal Gland Connection.
Khavinson's Peptide Bioregulation Theory
Khavinson's work extends beyond epitalon to a broader theory of peptide bioregulation, which proposes that short peptides (typically 2-4 amino acids) extracted from specific organs can restore youthful gene expression patterns in aging tissue. Khavinson et al. (2004) showed that specific dipeptides and tetrapeptides could promote cells to overcome the division limit, with the mechanism involving peptide-DNA interactions at the minor groove that modulate gene transcription.[7]
A 2021 systematic review by Khavinson et al. examined the evidence for peptide regulation of gene expression across multiple organ-specific peptide bioregulators.[8] The review found consistent evidence that short peptides modulate the expression of hundreds of genes in a tissue-specific manner, with effects on telomerase expression being one component of a broader geroprotective program.
This work has been criticized for lacking independent replication outside Russian research groups and for the absence of phase 3 clinical trials. The peptides are not FDA-regulated drugs and are primarily available through compounding pharmacies and research suppliers.
Other Peptides That Affect Telomeres
Epitalon is the most studied telomere-modulating peptide, but other peptides influence telomere biology through indirect mechanisms.
GHK-Cu (copper peptide) modulates the expression of over 4,000 genes, including genes involved in DNA repair and cellular senescence. Pickart et al. (2012) documented GHK-Cu's effects on oxidative stress and degenerative conditions, noting that the tripeptide upregulates antioxidant defense systems that protect telomeric DNA from oxidative damage.[9] Oxidative stress accelerates telomere shortening, so reducing it indirectly preserves telomere length. See GHK-Cu: The Copper Peptide That Modulates Over 4,000 Genes.
Growth hormone-releasing hormone (GHRH) antagonists have unexpected effects on telomerase. Banks et al. (2010) showed that a GHRH antagonist affected telomerase activity and oxidative stress markers in a cancer model, demonstrating that the GH/IGF-1 axis interacts with telomere maintenance pathways.[10] This connects the growth hormone signaling pathway, targeted by multiple peptide drugs, to the biology of cellular aging.
Mitochondrial-derived peptides (humanin, MOTS-c, SHLPs) interact with cellular senescence through metabolic pathways. Mendelsohn (2018) noted that some mitochondrial-derived peptides can exacerbate senescence in certain contexts, illustrating that the relationship between peptides and aging is not always beneficial.[11]
Modifiable Factors
What Shortens vs. What Protects Telomeres
Accelerates Shortening
Oxidative stress
GGG-rich telomere sequence is highly vulnerable to oxidative damage
Chronic psychological stress
Caregivers showed 9-17 years of extra telomere aging (Epel 2004)
Obesity
Chronic inflammation + increased oxidative burden
Smoking
Accelerates attrition via inflammation and oxidative damage
Physical inactivity
Reduced telomerase activity in immune cells
Preserves / Extends
Telomerase enzyme
Adds TTAGGG repeats — active in stem cells, silenced in most adult cells
Epitalon peptide
Reactivates telomerase in somatic cells + activates ALT pathway
Exercise
Associated with longer telomeres and increased telomerase activity
Stress reduction
Meditation linked to telomerase upregulation
Genetics (~70%)
Telomere length is highly heritable — sets your baseline
Source: Epel et al. (2004); Khavinson (2003); Sanchez (2026)
 View as image
View as imageWhat Shortens Telomeres Faster
Telomere attrition is not purely a function of cell division count. Multiple factors accelerate shortening beyond the baseline end-replication rate.
Oxidative stress damages telomeric DNA disproportionately because the GGG-rich telomere sequence is highly susceptible to oxidative modification. Each oxidative lesion in telomeric DNA may result in the loss of additional sequence during repair, effectively multiplying the per-division shortening rate.
Chronic psychological stress was linked to shorter telomeres by Epel et al. (2004) in a landmark study showing that mothers of chronically ill children had telomeres equivalent to 9-17 additional years of aging compared to controls. This finding connected the subjective experience of stress to measurable biological aging at the chromosomal level.
Obesity, smoking, and physical inactivity are all associated with shorter leukocyte telomere length in epidemiological studies. The mechanisms involve chronic inflammation (SASP-driven positive feedback), increased oxidative burden, and reduced telomerase activity in immune cells.
Heritability accounts for approximately 70% of telomere length variation, meaning genetic factors set the baseline and lifestyle factors modify the rate of decline. Some individuals are born with substantially longer telomeres and reach the critical threshold much later in life.
Telomere Length as a Biomarker: Strengths and Limitations
Commercial telomere testing is available, but interpreting the results requires understanding several caveats.
Most clinical telomere measurements assess leukocyte telomere length (LTL) from a blood sample. LTL correlates with telomere length in other tissues but is not identical. Different tissues age at different rates, and a blood-based measurement may not reflect telomere status in the specific tissue of clinical interest (brain, heart, liver).
The correlation between LTL and age-related disease risk is statistically significant but modest. Short telomeres are associated with increased risk of cardiovascular disease, type 2 diabetes, certain cancers, and all-cause mortality. However, the individual predictive value is low: two people with identical LTL may have vastly different health trajectories depending on other genetic and environmental factors.
For a complete analysis of what telomere testing can and cannot tell you, see Telomere Length as an Aging Biomarker: What It Tells Us and What It Doesn't.
The Cancer Tension
The relationship between telomeres, telomerase, and cancer creates a fundamental tension in anti-aging strategy. Short telomeres are a tumor-suppressive mechanism: they limit the number of times a cell can divide, preventing the accumulation of mutations that drive cancer. Reactivating telomerase to extend telomeres removes this brake.
Cancer cells overcome the telomere barrier by reactivating telomerase (85-90% of cancers) or using the ALT pathway (10-15%). The concern is that exogenous telomerase activation through peptides or small molecules could accelerate this process in pre-malignant cells.
The counterargument comes from animal data. Epitalon treatment in mice reduced spontaneous tumor incidence rather than increasing it.[5] One explanation is that telomerase activation in normal cells prevents the genomic instability caused by critically short telomeres, which itself is a driver of cancer initiation. By maintaining adequate telomere length, cells avoid the crisis period during which chromosomal fusions and translocations accumulate.
This remains an unresolved theoretical debate. The animal data is reassuring but limited. Long-term human safety data for telomerase-activating compounds does not exist at the scale needed for definitive conclusions.
Open Questions
Does telomerase activation translate to tissue-level rejuvenation? Extending telomeres in cultured fibroblasts is not the same as reversing aging in a living organism. Aging involves accumulated damage across multiple systems (protein aggregation, mitochondrial dysfunction, epigenetic drift, stem cell exhaustion), of which telomere shortening is one component. Fixing telomeres alone may not reverse the others.
What is the optimal telomere length? Extremely long telomeres are associated with increased cancer risk in some studies. The dose-response relationship between telomere extension and health benefit is unlikely to be linear, and the therapeutic window for telomerase activation may be narrower than the anti-aging community assumes.
Are epitalon's effects reproducible at scale? Most epitalon data comes from a single research group (Khavinson and colleagues) or from studies that build on their methodology. Independent replication by groups with no connection to the original researchers would strengthen the evidence considerably. The 2026 Sanchez et al. study represents one such independent confirmation.
How does epitalon compare to lifestyle interventions? Exercise, meditation, and dietary changes have all been associated with longer telomeres or increased telomerase activity in human studies. Whether epitalon provides benefits beyond what these lifestyle interventions achieve has not been tested in comparative trials.
What role do telomeres play in tissue-specific aging? Different tissues age at different rates, and telomere dynamics vary accordingly. Immune cells divide frequently and show rapid telomere attrition. Brain neurons rarely divide after development and maintain stable telomeres but are vulnerable to other aging mechanisms. Muscle satellite cells have intermediate division rates. A comprehensive anti-aging strategy would need to address telomere shortening in the tissues where it is rate-limiting while avoiding telomerase activation in tissues where cancer risk is highest. This tissue-specific complexity is rarely addressed in the consumer-facing telomere and anti-aging literature.
The Bottom Line
Telomere shortening is a fundamental mechanism of cellular aging, driven by the end-replication problem and accelerated by oxidative stress, inflammation, and psychological stress. The enzyme telomerase can reverse this shortening but is silenced in most adult cells. The tetrapeptide epitalon activates telomerase in human somatic cells and extends telomere length, with supporting evidence from lifespan studies in animals showing 12-31% increases in mean lifespan. A 2026 study confirmed epitalon acts through both telomerase upregulation and ALT pathway activation. However, large-scale human clinical trials are absent, independent replication is limited, and the cancer risk of telomerase activation remains a theoretical concern that animal data has so far not supported.
Sources & References
- 1RPEP-00833·Khavinson, V Kh et al. (2003). “Epithalon Peptide Activates Telomerase and Lengthens Telomeres in Human Cells.” Bulletin of experimental biology and medicine.Study breakdown →PubMed →↩
- 2RPEP-00738·Khavinson, V Kh et al. (2002). “Peptides and Ageing..” Neuro endocrinology letters.Study breakdown →PubMed →↩
- 3RPEP-16052·Sanchez-Martin, Veronica et al. (2026). “Epitalon increases telomere length in human cell lines through telomerase upregulation or ALT activity..” Biochemical and biophysical research communications.Study breakdown →PubMed →↩
- 4RPEP-00447·Anisimov, V N et al. (1998). “Pineal peptide preparation epithalamin increases the lifespan of fruit flies, mice and rats..” Mechanisms of ageing and development.Study breakdown →PubMed →↩
- 5RPEP-00788·Anisimov, V N et al. (2003). “Epitalon Peptide Extended Lifespan by 12% and Reduced Cancer in Aging Mice.” Biogerontology.Study breakdown →PubMed →↩
- 6RPEP-11540·Ivko, Olga M et al. (2025). “Overview of Epitalon-Highly Bioactive Pineal Tetrapeptide with Promising Properties..” Molecules (Basel.Study breakdown →PubMed →↩
- 7RPEP-00933·Khavinson, V Kh et al. (2004). “Epithalon Peptide Helps Human Cells Overcome the Hayflick Division Limit.” Bulletin of experimental biology and medicine.Study breakdown →PubMed →↩
- 8RPEP-05495·Khavinson, Vladimir Khatskelevich et al. (2021). “How Tiny Peptides Can Switch Genes On and Off: A Systematic Review.” Molecules (Basel.Study breakdown →PubMed →↩
- 9RPEP-02037·Pickart, Loren et al. (2012). “The human tripeptide GHK-Cu in prevention of oxidative stress and degenerative conditions of aging: implications for cognitive health..” Oxidative medicine and cellular longevity.Study breakdown →PubMed →↩
- 10RPEP-01584·Banks, William A et al. (2010). “Blocking Growth Hormone-Releasing Hormone Affects Telomerase, Aging, and Oxidative Stress in Mice.” Proceedings of the National Academy of Sciences of the United States of America.Study breakdown →PubMed →↩
- 11RPEP-03808·Mendelsohn, Andrew R et al. (2018). “Mitochondrial-Derived Peptides Exacerbate Senescence..” Rejuvenation research.Study breakdown →PubMed →↩