Defensins in Your Lungs: Airway Defense Peptides
Lung Defense Peptides
6+ defensin types in airways
Human airways produce at least six defensin peptides that kill pathogens and regulate immune responses, forming a critical layer of innate defense that operates before the adaptive immune system engages.
Cole et al., Am J Resp Med, 2002
Cole et al., Am J Resp Med, 2002
If you only read one thing
Every breath pulls germs into your lungs, and mostly you do not get sick. Part of the reason is that your airway cells are coating their own surface with tiny peptide weapons called defensins. One of them is always on, like a porch light. Others switch on within hours of a bacteria or virus arriving. Neutrophils — the first immune cells to rush to trouble — carry huge stockpiles of a different defensin class and dump them on the infection site. This whole system is well-mapped in the lab. There is still no defensin drug on the market for lung infections, but the biology is one of the strongest reasons you can breathe in a forest full of microbes and be fine.
Every breath brings potential pathogens into the lungs. Bacteria, fungi, and viruses all ride airflow into the respiratory tract, yet lung infections are relatively rare in healthy people. Part of the reason is a class of small antimicrobial peptides called defensins, which are produced by airway epithelial cells and neutrophils and secreted into the thin layer of fluid that lines the respiratory surfaces. These peptides kill microorganisms directly, recruit immune cells, and help shape the inflammatory response. A 2002 review in the American Journal of Respiratory Medicine catalogued the defensins present in human lung fluid and concluded they represent "key elements in this innate immune system."[1] This article examines which defensins operate in the lungs, how they work, what happens when they fail, and what makes them different from the body's other antimicrobial systems. For context on how defensins fit into the broader antimicrobial peptide landscape, see Antimicrobial Peptides as Alternatives to Antibiotics: Can They Solve Resistance?.
Key Takeaways
- Human airways produce at least 4 beta-defensins (HBD-1 through HBD-4) from epithelial cells and receive alpha-defensins (HNP-1 through HNP-4) from infiltrating neutrophils
- HBD-1 is produced constitutively (always on), while HBD-2 and HBD-3 are induced by bacterial products and pro-inflammatory cytokines like IL-1beta (Singh et al., 1998)
- In cystic fibrosis patients, decreased beta-defensin levels in bronchoalveolar lavage fluid correlate with advanced lung disease, suggesting a secondary innate defense defect (Chen et al., 2004)
- Beyond direct killing, defensins recruit inflammatory cells, activate dendritic cells, and can serve as vaccine adjuvants by bridging innate and adaptive immunity (Kim et al., 2018)
- Alpha-defensins at high concentrations in AATD patients paradoxically increased bacterial binding to macrophages without improving killing, potentially worsening chronic airway infection (Lee et al., 2025)
- Defensins have broad-spectrum antiviral activity, including against respiratory viruses, through mechanisms ranging from direct viral binding to interference with host cell entry (Zupin et al., 2022)
What Are Defensins?
Defensins are small (3-5 kDa), cationic, cysteine-rich peptides that form part of the innate immune system. They are among the most ancient antimicrobial molecules in biology, with homologs found in organisms from plants to insects to mammals, suggesting they evolved over 500 million years ago as a fundamental defense strategy. In humans, defensins are divided into two subfamilies based on the spacing of their three characteristic disulfide bonds: alpha-defensins and beta-defensins. A third subfamily (theta-defensins) exists in some primates but is not produced as functional peptides in humans due to a premature stop codon in the gene.[1]
The three disulfide bonds give defensins a compact, stable structure that resists degradation by proteases in the harsh environment of airway surface liquid, the gastrointestinal tract, and inflammatory exudates. This structural stability is essential for their function: a peptide that is rapidly degraded cannot maintain antimicrobial concentrations at mucosal surfaces.
Alpha-defensins (also called human neutrophil peptides, HNP-1 through HNP-4) are stored in the azurophilic granules of neutrophils. They are not produced by airway epithelial cells but arrive in the lungs when neutrophils are recruited to sites of infection or inflammation. A fifth and sixth alpha-defensin (HD-5 and HD-6) are produced by intestinal Paneth cells and do not play a direct role in lung defense. For a detailed look at alpha-defensin biology beyond the lungs, see Alpha-Defensins: The Neutrophil Peptides That Kill Bacteria on Contact.
Beta-defensins (HBD-1 through HBD-4) are produced by airway epithelial cells and are the defensins most directly associated with mucosal lung defense. Twenty-six human DEFB genes have been identified, but only HBD-1 through HBD-4 are confirmed to be secreted by the respiratory tract.
How Airway Epithelial Cells Produce Defensins
Airway defensin regulation
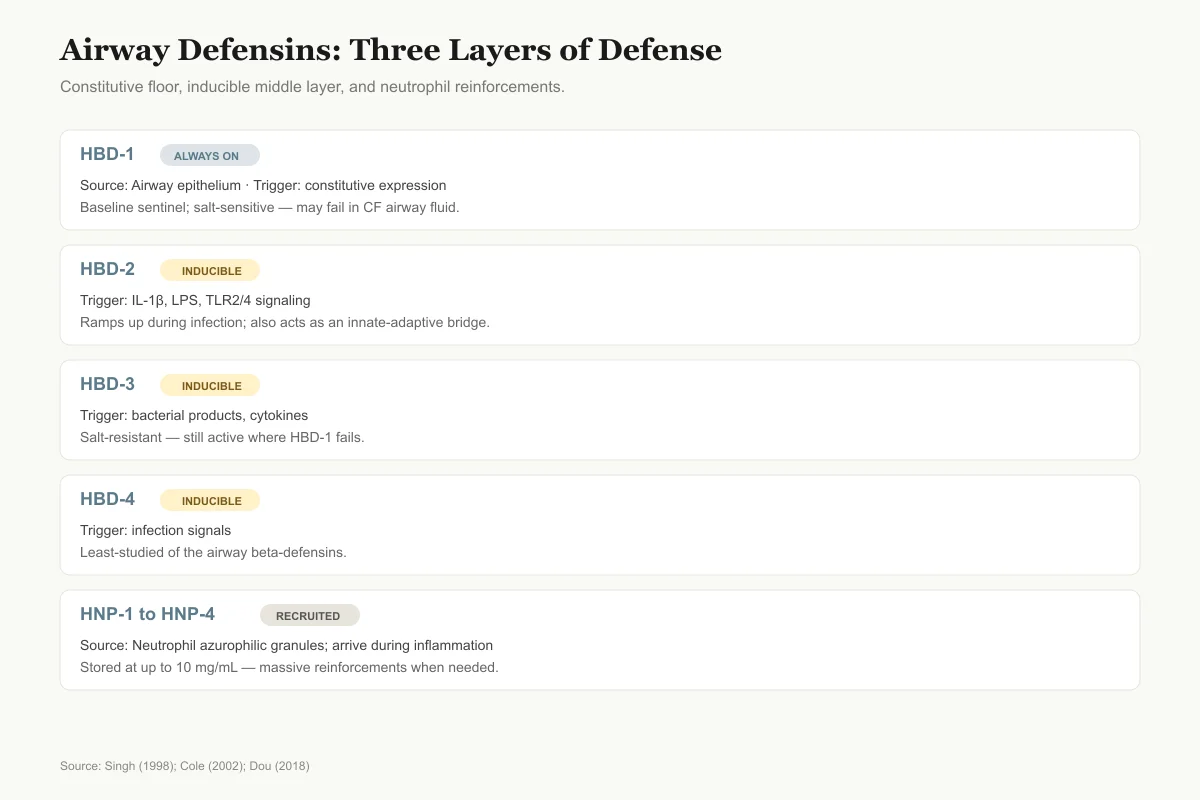
Three Layers of Defense, Three Different Triggers
A constitutive floor, an inducible middle layer, and neutrophil reinforcements arriving when things get serious.
HBD-1
Always onSource cell
Airway epithelium
What turns it on
Always on
Notable trait
Baseline sentinel; salt-sensitive
HBD-2
InducibleSource cell
Airway epithelium
What turns it on
IL-1β, LPS, TLR2/4
Notable trait
Ramps up during infection
HBD-3
InducibleSource cell
Airway epithelium
What turns it on
Bacterial products, cytokines
Notable trait
Salt-resistant; CF-relevant
HBD-4
InducibleSource cell
Airway epithelium
What turns it on
Infection signals
Notable trait
Least-studied airway beta-defensin
HNP-1 to HNP-4
RecruitedSource cell
Neutrophils (recruited)
What turns it on
Neutrophil degranulation at site of inflammation
Notable trait
Stored at up to 10 mg/mL in granules
Only HBD-1 is always on. The rest wait for a signal. That layered on-demand system is part of why a healthy airway can clear inhaled pathogens without runaway inflammation.
Source: Singh et al. (1998); Cole et al. (2002); Dou et al. (2018)
 View as image
View as imageThe foundational study on airway beta-defensin production was published by Singh et al. in 1998 in the Proceedings of the National Academy of Sciences. They demonstrated that HBD-1 and HBD-2 mRNAs are expressed in both surface and submucosal gland epithelia from non-cystic fibrosis and cystic fibrosis patients.[2]
A critical distinction emerged from this work. HBD-1 is constitutively expressed, meaning it is always being produced regardless of whether an infection is present. It serves as a standing sentinel in the airway fluid. HBD-2, by contrast, is inducible: its expression is upregulated by the pro-inflammatory cytokine interleukin-1beta (IL-1beta) and by direct contact with bacterial products like lipopolysaccharide. This two-tiered system provides both constant baseline protection (HBD-1) and amplified response during active infection (HBD-2).
The production pathway involves Toll-like receptor (TLR) signaling. Dou et al. (2018) demonstrated that TLR2/4-mediated NF-kappaB pathway activation, combined with histone modification, regulates beta-defensin expression in response to Staphylococcus aureus infection. This epigenetic component means that defensin production is not simply an on/off switch but a tunable response that can be modulated by the chromatin state of defensin gene promoters.[3]
How Defensins Kill Pathogens
Mechanism Comparison
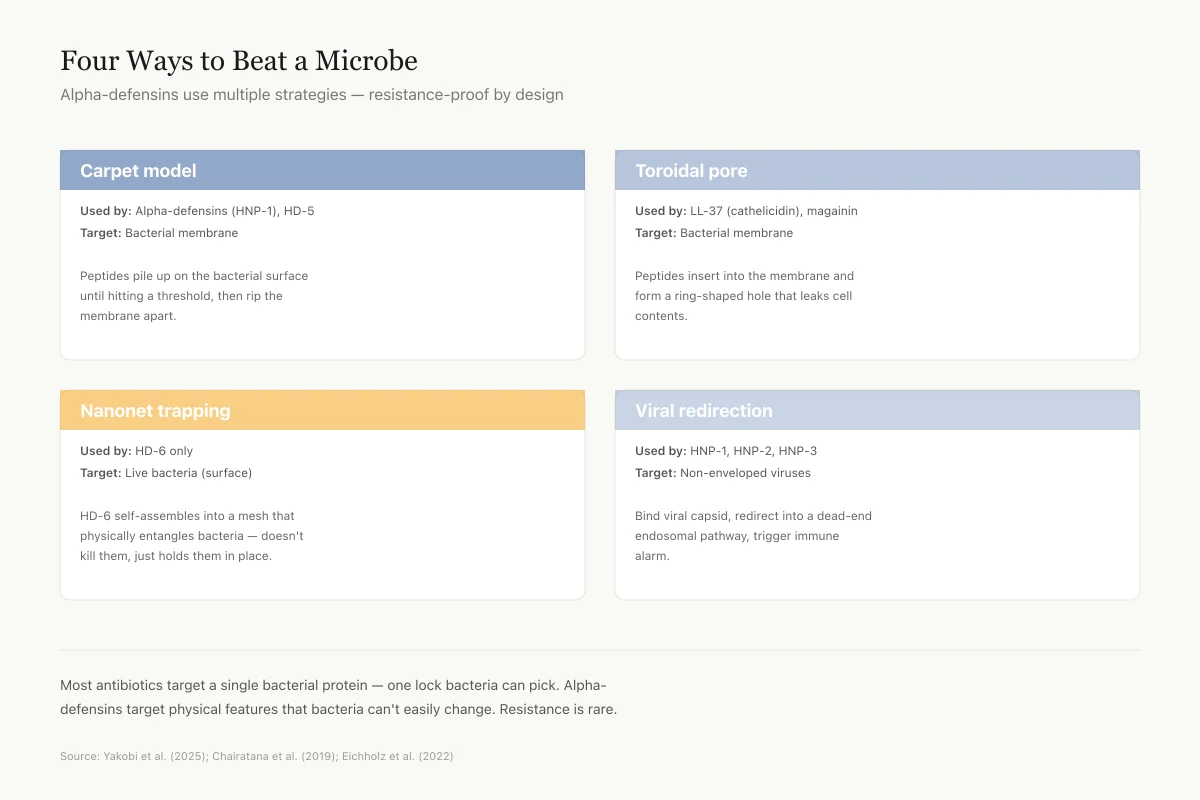
Four Different Ways to Beat a Microbe
Alpha-defensins use multiple strategies — resistance-proof by design
Carpet model
Used by: Alpha-defensins (HNP-1), HD-5
Target: Bacterial membrane
Peptides pile up on the bacterial surface until they hit a threshold, then rip the membrane apart.
Toroidal pore
Used by: LL-37 (cathelicidin), magainin
Target: Bacterial membrane
Peptides insert into the membrane and form a ring-shaped hole that leaks cell contents out.
Nanonet trapping
Used by: HD-6 only
Target: Live bacteria (surface)
HD-6 self-assembles into a mesh-like net that physically entangles bacteria without killing them directly.
Viral redirection
Used by: HNP-1, HNP-2, HNP-3
Target: Non-enveloped viruses (adenovirus)
Bind to viral capsid, redirect the virus into a dead-end endosomal pathway, trigger immune alarm.
Most antibiotics target a single bacterial protein — one lock bacteria can eventually pick. Alpha-defensins target physical features (charge, membrane lipids, viral capsid shape) that bacteria and viruses can't easily change. That's why resistance is rare.
Source: Yakobi et al. (2025); Chairatana et al. (2019); Eichholz et al. (2022)
 View as image
View as imageDefensins kill microorganisms primarily through membrane disruption. Their cationic (positively charged) surfaces are attracted to the negatively charged membranes of bacteria and fungi. Upon binding, they insert into the lipid bilayer and form pores or channels, causing membrane permeabilization and cell death. This mechanism is difficult for bacteria to develop resistance against because it would require fundamental changes to membrane composition, which is one reason defensins have remained effective antimicrobial agents across hundreds of millions of years of evolution.
The killing spectrum is broad. Defensins are active against gram-positive bacteria (including Staphylococcus aureus and Streptococcus pneumoniae, two major respiratory pathogens), gram-negative bacteria (including Pseudomonas aeruginosa, the most problematic pathogen in cystic fibrosis), fungi including Candida and Aspergillus species, and enveloped viruses. The specificity for microbial membranes over host cell membranes derives from differences in lipid composition: mammalian cell membranes are rich in cholesterol and neutral phospholipids, while bacterial membranes contain more negatively charged phospholipids that attract the cationic defensins.[4]
The antimicrobial concentrations required for killing vary by defensin type and target organism. Alpha-defensins typically kill bacteria at concentrations of 1-10 microg/mL, while beta-defensins are effective at similar or slightly higher concentrations. These concentrations are achievable in the thin airway surface liquid where defensins are concentrated, though bulk measurements of whole BAL fluid may underestimate local concentrations at the epithelial surface.
Salt Sensitivity: A Critical Limitation
A complication specific to lung defense is salt sensitivity. HBD-1 and some alpha-defensins lose antimicrobial activity at the salt concentrations found in airway surface liquid. This is directly relevant to cystic fibrosis, where mutations in the CFTR chloride channel alter the ionic composition of airway fluid. The hypothesis that defensin inactivation by abnormal salt concentrations contributes to CF lung infections has been debated since the late 1990s and remains an active area of investigation.[2]
Not all defensins are equally salt-sensitive. HBD-3 retains antimicrobial activity at higher salt concentrations than HBD-1 or HBD-2, which is one reason it has received attention as a potential therapeutic agent. The structural basis for this difference lies in the distribution of charged residues: HBD-3 has a higher net positive charge that maintains electrostatic attraction to bacterial membranes even in higher ionic strength environments. This salt resistance makes HBD-3 particularly relevant to the CF airway, where elevated sodium chloride concentrations would inactivate salt-sensitive defensins.
Beyond Killing: Immunomodulatory Functions
The initial characterization of defensins focused on their antimicrobial properties. More recent research has revealed that defensins also function as signaling molecules that coordinate broader immune responses.
Chemotaxis and Immune Cell Recruitment
Defensins recruit immune cells to sites of infection. Alpha-defensins attract monocytes and T cells. Beta-defensins, particularly HBD-2, recruit dendritic cells and memory T cells through interaction with the chemokine receptor CCR6. This chemotactic activity means defensins do not just kill pathogens at the site of infection; they also summon reinforcements from the adaptive immune system.[4]
Antiviral Activity
Zupin et al. (2022) published a review examining defensins' antiviral properties, emphasizing their role in respiratory tract defense. The review catalogued mechanisms including direct viral binding, interference with viral entry into host cells, and modulation of host cell signaling pathways that viruses exploit for replication. Human defensins are expressed at the level of the respiratory tract where they represent the first line of defense against respiratory viruses.[5]
Vaccine Adjuvant Potential
Kim et al. (2018) demonstrated that HBD-2 plays a regulatory role in innate antiviral immunity and can potentiate antigen-specific immune responses. In experimental models, HBD-2 enhanced the immunogenicity of co-administered antigens, functioning as a natural adjuvant. The enhancement occurred through activation of innate immune pathways that prime the adaptive immune response. This has implications for vaccine design: a molecule already present in the airway could theoretically be harnessed to boost mucosal immune responses to inhaled vaccines, potentially eliminating the need for synthetic adjuvants that can cause local irritation or systemic inflammatory side effects.[6]
Defensins in Lung Disease
Genetic Variation
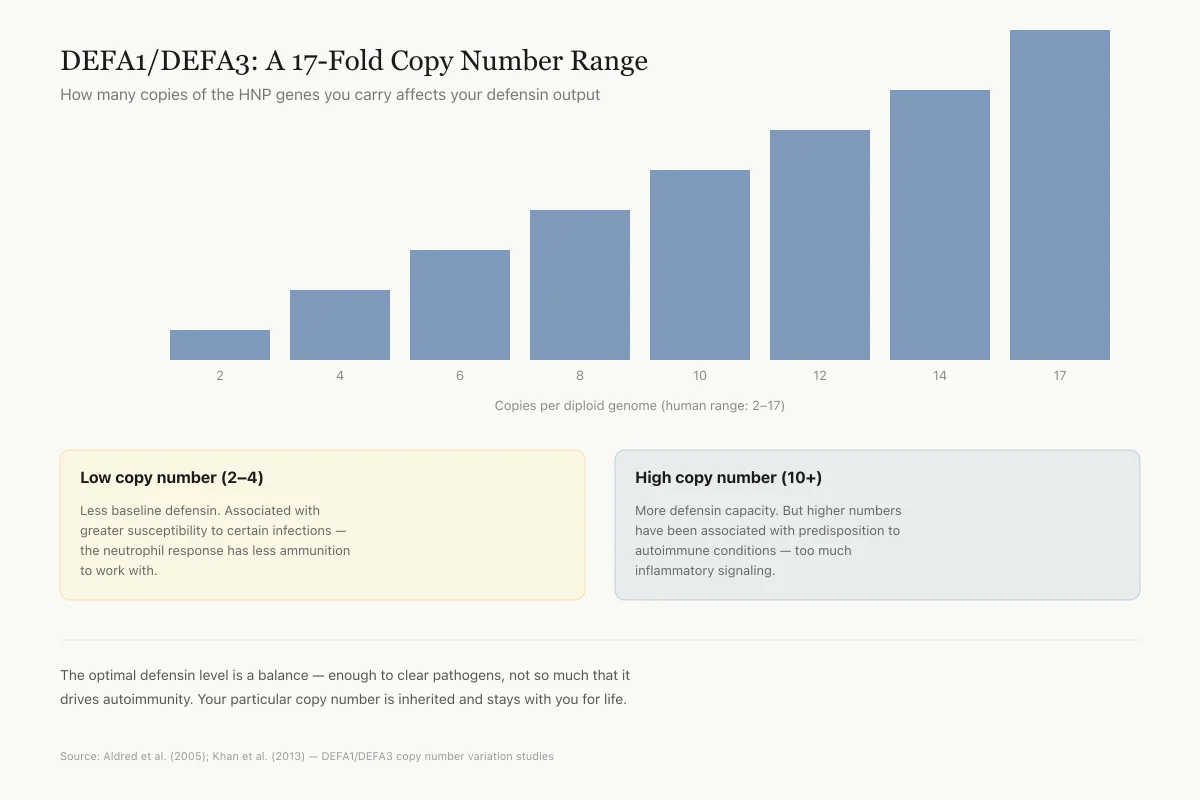
DEFA1/DEFA3: A 17-Fold Copy Number Range
How many copies of the HNP genes you carry affects your defensin output
Copies per diploid genome (human range: 2–17)
Low copy number (2–4)
Less baseline defensin. Associated with greater susceptibility to certain infections — the neutrophil response has less ammunition to work with.
High copy number (10+)
More defensin capacity. But higher numbers have been associated with predisposition to autoimmune conditions — too much inflammatory signaling.
This is one of the most dramatic examples of structural variation in the human genome. The optimal defensin level is a balance — enough to clear pathogens, not so much that it drives autoimmunity. Your particular copy number is inherited and stays with you for life.
Source: Aldred et al. (2005); Khan et al. (2013) — DEFA1/DEFA3 copy number variation studies
 View as image
View as imageCystic Fibrosis
Chen et al. (2004) measured HBD-1, HBD-2, and LL-37 concentrations in bronchoalveolar lavage (BAL) fluid from CF patients with mild lung disease. They found that LL-37/hCAP-18 levels were associated with bronchial inflammation and disease severity, while decreased levels of beta-defensins in advanced lung disease likely contribute to a secondary defect in local host defense.[7]
This finding shifts the CF defensin story beyond the salt sensitivity hypothesis. Even if defensin activity is reduced by altered ionic composition (the original hypothesis), the actual concentration of defensin peptides also decreases as disease progresses. The result is a double deficit: less defensin produced and reduced activity of what is produced. Whether this contributes to the progressive bacterial colonization characteristic of CF (particularly with Pseudomonas aeruginosa and Staphylococcus aureus) remains an active question. Biswas et al. (2021) detailed the molecular mechanisms by which these two organisms interact and establish chronic infections in CF airways, including their interactions with host defense peptides.[8]
Alpha-1 Antitrypsin Deficiency
A 2025 study by Lee et al. revealed a paradoxical role for alpha-defensins in alpha-1 antitrypsin deficiency (AATD). AATD patients have high levels of free alpha-defensins in their airways due to chronic neutrophilic inflammation. The study found that these elevated alpha-defensins increased NTHi (non-typeable Haemophilus influenzae) binding to macrophages but did not enhance bacterial engulfment or killing. This means the defensins made bacteria stick to immune cells without improving clearance, potentially enhancing rather than reducing airway inflammation.[9]
This finding illustrates a broader principle: defensins at physiological concentrations have protective effects, but at pathologically elevated concentrations they can become pro-inflammatory and paradoxically harmful. The dose-response relationship is not linear, and more defensin does not always mean better defense. The AATD data reinforces that defensins must be studied in specific disease contexts rather than assumed to be uniformly protective. In a healthy airway, alpha-defensins help clear bacteria. In an AATD airway, the same molecules may contribute to the cycle of chronic infection and inflammation that drives disease progression.
Interstitial Lung Disease
Sakamoto et al. (2018) found elevated alpha-defensin levels in both plasma and BAL fluid from patients with myositis-associated interstitial lung disease (ILD). The elevations correlated with neutrophil activation markers, suggesting that defensin levels in BAL fluid may serve as biomarkers for disease activity in ILD. This represents a diagnostic rather than therapeutic application of defensin biology. If validated in larger cohorts, defensin levels in BAL fluid could help distinguish active from quiescent ILD, guide treatment intensity, and monitor response to immunosuppressive therapy. The advantage of defensins as biomarkers is their relative specificity for neutrophilic inflammation, which helps differentiate neutrophil-driven from lymphocyte-driven or fibrotic processes in complex lung diseases.[10]
COPD and the Defensin Paradox
Chronic obstructive pulmonary disease (COPD) presents a different defensin story than cystic fibrosis. In COPD, the problem is not defensin deficiency but potential defensin excess and dysregulation. Cigarette smoke exposure triggers chronic neutrophilic inflammation, flooding the airways with alpha-defensins from degranulating neutrophils. Aarbiou et al. (2002) reviewed evidence that alpha-defensins, while antimicrobial at low concentrations, can amplify cigarette smoke-induced and infection-induced inflammatory reactions at high concentrations, leading to epithelial damage and accelerated lung injury.[4]
The COPD defensin problem mirrors the AATD findings from Lee et al.: too much defensin in the wrong context can be harmful. In healthy airways, defensin concentrations are tightly regulated. The constitutive expression of HBD-1 provides low-level baseline defense. Inducible HBD-2 and HBD-3 ramp up during infection and ramp down as the infection resolves. Alpha-defensins arrive with recruited neutrophils and disappear when inflammation resolves. In COPD, chronic inflammation means continuously elevated alpha-defensin levels, turning a defensive molecule into a contributor to tissue damage.
This dual nature of defensins (protective at physiological levels, harmful at pathological levels) has implications for any therapeutic strategy that aims to increase defensin concentrations in the airway. Simply boosting defensin levels without addressing the inflammatory context could worsen disease rather than improve it.
Defensins and LL-37: Complementary Systems
Defensins do not operate alone in the airway. They work alongside another major antimicrobial peptide family: the cathelicidins, represented in humans by LL-37 (also designated hCAP-18). While defensins and LL-37 both kill pathogens through membrane disruption, they have different salt sensitivities, different expression patterns, and different immunomodulatory profiles. LL-37 is more salt-resistant than most defensins and has stronger anti-biofilm activity, making it particularly relevant to chronic lung infections where bacteria form protected biofilms on airway surfaces. For a dedicated analysis, see LL-37 in Respiratory Immunity: Your Airway's Natural Antibiotic.
Chen et al. (2004) measured both defensins and LL-37 in CF BAL fluid, finding that the two peptide classes show different patterns of dysregulation. LL-37 levels correlated with inflammation severity, while beta-defensin levels decreased with disease progression.[7] This dissociation suggests that defensins and cathelicidins are regulated by different pathways and that their contributions to airway defense are complementary rather than redundant.
The practical implication is that studying one antimicrobial peptide class in isolation gives an incomplete picture of airway innate defense. The defensin system and the cathelicidin system together provide overlapping but distinct protective coverage, and disease states may affect one system without equally affecting the other.
Defensins and Lung Surfactant
The lung's air-liquid interface is covered by pulmonary surfactant, a complex mixture of lipids and proteins that reduces surface tension and prevents alveolar collapse. Defensins must operate within this surfactant layer to reach pathogens, and the interaction between defensins and surfactant components affects their antimicrobial activity.
Souza et al. (2019) used molecular dynamics simulations to study how HBD-3 encapsulated with polyethylene glycol permeates through lung surfactant models. They found that HBD-3 can penetrate surfactant layers at physiological surface tensions but interacts with the lipid components in ways that affect its distribution and availability. This work is relevant to therapeutic applications: delivering defensin-based drugs to the lower airways requires them to cross the surfactant barrier while maintaining antimicrobial activity.[11]
The interaction between defensins and surfactant also has implications for surfactant replacement therapy in premature infants, whose lungs are deficient in both surfactant and innate immune peptides. Exogenous surfactant preparations used in neonatal intensive care units do not contain defensins or other antimicrobial peptides, which may partly explain why premature infants remain vulnerable to respiratory infections even after surfactant replacement restores mechanical lung function.
Vitamin D and Defensin Expression
Bleakley et al. (2021) reviewed evidence that vitamin D modulates the innate immune response to pediatric respiratory pathogens, including through regulation of defensin and cathelicidin expression. Vitamin D receptor activation upregulates several antimicrobial peptides in respiratory epithelial cells, providing a molecular link between vitamin D status and susceptibility to respiratory infections.[12]
This connection has been proposed as one mechanism behind the epidemiological association between low vitamin D levels and increased respiratory infection risk. The molecular pathway is plausible: vitamin D response elements exist in the promoter regions of several defensin and cathelicidin genes, and 1,25-dihydroxyvitamin D3 (the active form) directly increases transcription of these genes in airway epithelial cell culture models.
However, clinical trials of vitamin D supplementation for respiratory infection prevention have produced mixed results. A possible explanation is that the relationship between systemic vitamin D levels, local vitamin D metabolism in airway epithelial cells, defensin gene transcription, defensin peptide secretion, and actual antimicrobial activity in airway fluid involves multiple steps where the signal can be attenuated. Supplementation that raises serum vitamin D levels may not proportionally increase defensin production at the airway surface, particularly if other regulatory mechanisms (cytokine signals, epigenetic modifications, pathogen exposure) are the rate-limiting factors.
What the Evidence Shows and What It Does Not
The defensin system in the lungs is well characterized at the molecular and cellular level. We know which defensins are produced, which cells produce them, what triggers their expression, and how they kill pathogens. This mechanistic understanding is substantially more complete than for many other peptide systems.
What remains less clear is the quantitative contribution of defensins to overall lung defense in living human airways. Most antimicrobial activity measurements come from in vitro assays where defensin concentrations, salt levels, and pathogen loads are controlled. In the living airway, defensins operate alongside dozens of other antimicrobial molecules (lysozyme, lactoferrin, collectins, LL-37) in a complex fluid with variable ionic composition. Isolating the specific contribution of any one peptide class to overall airway defense is methodologically difficult, and knockout mouse studies (where individual defensin genes are deleted) have sometimes shown smaller effects than expected from in vitro potency, suggesting substantial redundancy in the antimicrobial peptide system.
The therapeutic potential of defensins for respiratory infections remains largely theoretical. No defensin-based drug has reached clinical trials for lung disease. The salt sensitivity of some defensins, the complexity of airway delivery, and the challenge of producing correctly folded synthetic defensins at therapeutic scales are practical barriers.
Two near-term applications show more promise than direct therapeutics. First, using defensin levels as biomarkers for disease activity in conditions like ILD and COPD, where alpha-defensin concentrations in BAL fluid correlate with neutrophilic inflammation and disease progression. Second, exploiting defensins' adjuvant properties for inhaled vaccine development, where HBD-2's demonstrated ability to enhance antigen-specific immune responses could improve mucosal vaccine efficacy without requiring systemic adjuvants.
The Bottom Line
Lung defensins are a well-characterized family of antimicrobial peptides that provide front-line airway defense through direct pathogen killing, immune cell recruitment, and modulation of inflammatory responses. Alpha-defensins arrive from neutrophils; beta-defensins are produced by airway epithelial cells, with HBD-1 constitutively expressed and HBD-2/3 induced by infection. In disease states like cystic fibrosis and AATD, defensin dysfunction or dysregulation contributes to chronic infection susceptibility. No defensin-based therapeutic has reached clinical trials for lung disease, but diagnostic applications as biomarkers show promise.
Sources & References
- 1RPEP-00720·Cole, Alexander M et al. (2002). “Defensins in the Lungs: How Natural Antimicrobial Peptides Protect Your Airways and Could Become Drugs.” American journal of respiratory medicine : drugs.Study breakdown →PubMed →↩
- 2RPEP-00495·Singh, P K et al. (1998). “Production of beta-defensins by human airway epithelia..” Proceedings of the National Academy of Sciences of the United States of America.Study breakdown →PubMed →↩
- 3RPEP-03652·Dou, Xiujing et al. (2018). “Boosting Your Gut's Natural Antibiotic Peptides Without Causing Inflammation.” Food & nutrition research.Study breakdown →PubMed →↩
- 4RPEP-00710·Aarbiou, Jamil et al. (2002). “Role of defensins in inflammatory lung disease..” Annals of medicine.Study breakdown →PubMed →↩
- 5RPEP-06660·Zupin, Luisa et al. (2022). “Human Defensins from Antivirals to Vaccine Adjuvants: Rediscovery of the Innate Immunity Arsenal..” Protein and peptide letters.Study breakdown →PubMed →↩
- 6RPEP-03752·Kim, Ju et al. (2018). “Human β-defensin 2 plays a regulatory role in innate antiviral immunity and is capable of potentiating the induction of antigen-specific immunity..” Virology journal.Study breakdown →PubMed →↩
- 7RPEP-00892·Chen, Christiane I-U et al. (2004). “Beta-defensins and LL-37 in bronchoalveolar lavage fluid of patients with cystic fibrosis..” Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society.Study breakdown →PubMed →↩
- 8RPEP-05286·Biswas, Lalitha et al. (2021). “Molecular Mechanisms of Staphylococcus and Pseudomonas Interactions in Cystic Fibrosis..” Frontiers in cellular and infection microbiology.Study breakdown →PubMed →↩
- 9RPEP-12018·Lee, Jungnam et al. (2025). “Alpha-defensins increase NTHi binding but not engulfment by the macrophages enhancing airway inflammation in Alpha-1 antitrypsin deficiency..” Frontiers in immunology.Study breakdown →PubMed →↩
- 10RPEP-03881·Sakamoto, Noriho et al. (2018). “Alpha-Defensin Levels Are Elevated in Patients With Myositis-Related Lung Disease.” BMC pulmonary medicine.Study breakdown →PubMed →↩
- 11RPEP-04492·Souza, F R et al. (2019). “Permeation of beta-defensin-3 encapsulated with polyethylene glycol in lung surfactant models at air-water interface..” Colloids and surfaces. B.Study breakdown →PubMed →↩
- 12RPEP-05287·Bleakley, Amy S et al. (2021). “Vitamin D Modulation of the Innate Immune Response to Paediatric Respiratory Pathogens Associated with Acute Lower Respiratory Infections..” Nutrients.Study breakdown →PubMed →↩