Defensins Against Influenza and COVID-19
Airway Antimicrobial Peptides
Submicromolar spike binding
Human neutrophil defensin HNP1 bound the SARS-CoV-2 spike protein with submicromolar affinity, over 20-fold stronger than its binding to serum albumin, and blocked viral infection in cell culture.
Kudryashova et al., Journal of Molecular Biology, 2022
Kudryashova et al., Journal of Molecular Biology, 2022
If you only read one thing
Your airway is coated in tiny antimicrobial peptides called defensins. They work like bouncers — always on, and they get louder the moment a virus shows up. Against COVID, one defensin (HNP1) grips the spike protein and bends it out of shape. Another (HD5) sticks to the ACE2 doorway on your cells so the virus cannot get in. Against flu they clump virus particles together so fewer can reach your cells. This is well-proven in lab dishes. None of it has been turned into a defensin-based drug or nasal spray you can buy, but it is one of the more promising directions for future antiviral research.
Before a respiratory virus reaches the first cell it can infect, it must pass through a gauntlet of antimicrobial peptides coating the airway surface. Defensins are the most abundant of these peptides. Human airways constitutively express beta-defensin 1 (HBD-1) and rapidly upregulate beta-defensin 2 (HBD-2) during infection, while neutrophils recruited to sites of inflammation release alpha-defensins (HNP1-4) in massive quantities. Together, these peptides form a chemical barrier that can neutralize influenza viruses, SARS-CoV-2, and other respiratory pathogens before adaptive immunity even activates. Wilson et al. (2013) reviewed the antiviral mechanisms of human defensins in the Journal of Molecular Biology, identifying direct viral disruption, receptor blocking, fusion inhibition, and immune modulation as four distinct modes of antiviral action.[1] Since then, the COVID-19 pandemic has generated a surge of research connecting defensin levels to disease severity and demonstrating that defensins can block SARS-CoV-2 infection in laboratory models. This article covers the full evidence landscape of defensin antiviral activity in respiratory infections. For related topics, see the cluster articles on cathelicidins and respiratory viruses and how antimicrobial peptides defend your lungs.
Key Takeaways
- Human neutrophil defensin HNP1 bound the SARS-CoV-2 spike protein with submicromolar affinity, destabilized it, and blocked both pseudovirus and authentic SARS-CoV-2 infection, including the B.1.1.7 and P.1 variants (Kudryashova et al., J Mol Biol, 2022).[2]
- Multiple human defensins (HNP1-3, HD5, HBD-2, HBD-3) blocked SARS-CoV-2 pseudovirus entry into cells, with HD5 specifically cloaking the ACE2 receptor to prevent spike protein binding (Xu et al., Viruses, 2021).[3]
- Human beta-defensin 2 potentiated antiviral innate immunity by enhancing interferon and ISG expression beyond its direct antimicrobial effects (Kim et al., Virology Journal, 2018).[4]
- SARS-CoV-2 infection downregulated defensin gene expression in patients, suggesting the virus actively suppresses innate peptide defenses (Idris et al., Acta Virologica, 2022).[10]
- Plasma alpha-defensin-1 levels correlated with COVID-19 severity across a multi-omic analysis, and DEFA1 inhibited SARS-CoV-2 pseudovirus infection in vitro (Qian et al., J Med Virol, 2023).[9]
- Defensins exert evolutionary pressure on viruses, with evidence that viral populations evolve resistance to defensin-mediated neutralization over time (Diaz & Smith, PLoS Pathogens, 2020).[6]
The defensin family: three classes, one mission
Humans produce three structural classes of defensins, each with distinct tissue distributions and modes of action.
Alpha-defensins include six members. HNP1-4 (human neutrophil peptides) are stored in neutrophil azurophilic granules at extraordinarily high concentrations, up to 10 mg/mL, and released during degranulation at sites of infection. HD5 and HD6 (human defensins 5 and 6) are expressed by Paneth cells in the small intestine. All alpha-defensins are 29 to 35 amino acids with three intramolecular disulfide bonds in a conserved pairing pattern (Cys1-Cys6, Cys2-Cys4, Cys3-Cys5).[1]
Beta-defensins are expressed primarily by epithelial cells, including the airway epithelium that represents the first point of contact for respiratory viruses. HBD-1 is constitutively expressed, providing a baseline defense layer even in the absence of infection. HBD-2, HBD-3, and HBD-4 are inducible, with expression upregulated by inflammatory cytokines, bacterial products, and viral infection. Beta-defensins have a different disulfide connectivity (Cys1-Cys5, Cys2-Cys4, Cys3-Cys6) but adopt a similar three-dimensional fold to alpha-defensins.[1]
Theta-defensins are the only cyclic defensins in mammals, formed by the head-to-tail ligation of two truncated alpha-defensin precursors. Humans carry theta-defensin genes (DEFT), but they contain a premature stop codon that prevents expression. Old World primates like rhesus macaques produce functional theta-defensins called retrocyclins, which have potent antiviral activity. Synthetic human retrocyclins, engineered by correcting the stop codon, have been studied as potential antiviral therapeutics.[2]
Four mechanisms of antiviral action
Mechanism Comparison
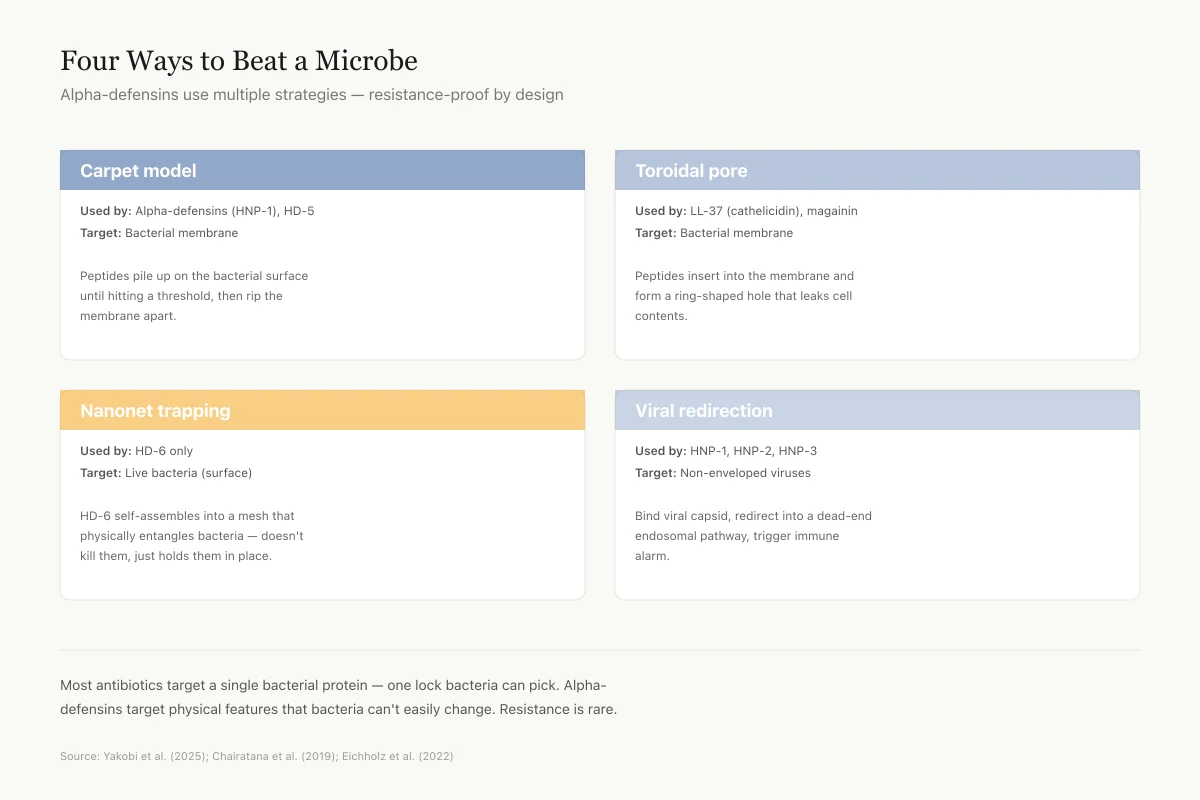
Four Different Ways to Beat a Microbe
Alpha-defensins use multiple strategies — resistance-proof by design
Carpet model
Used by: Alpha-defensins (HNP-1), HD-5
Target: Bacterial membrane
Peptides pile up on the bacterial surface until they hit a threshold, then rip the membrane apart.
Toroidal pore
Used by: LL-37 (cathelicidin), magainin
Target: Bacterial membrane
Peptides insert into the membrane and form a ring-shaped hole that leaks cell contents out.
Nanonet trapping
Used by: HD-6 only
Target: Live bacteria (surface)
HD-6 self-assembles into a mesh-like net that physically entangles bacteria without killing them directly.
Viral redirection
Used by: HNP-1, HNP-2, HNP-3
Target: Non-enveloped viruses (adenovirus)
Bind to viral capsid, redirect the virus into a dead-end endosomal pathway, trigger immune alarm.
Most antibiotics target a single bacterial protein — one lock bacteria can eventually pick. Alpha-defensins target physical features (charge, membrane lipids, viral capsid shape) that bacteria and viruses can't easily change. That's why resistance is rare.
Source: Yakobi et al. (2025); Chairatana et al. (2019); Eichholz et al. (2022)
 View as image
View as imageWilson et al. (2013) categorized the antiviral mechanisms of human defensins into four modes, each supported by experimental evidence across multiple virus families.[1]
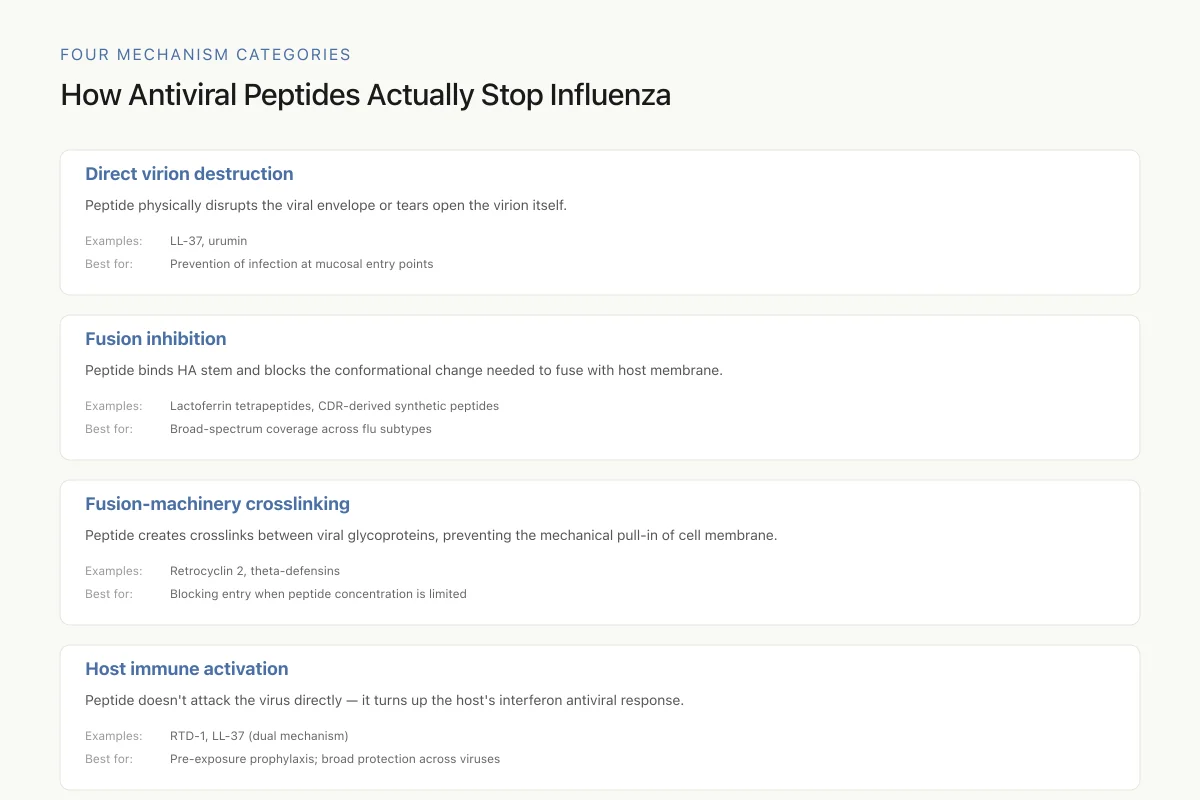
Direct viral neutralization. Defensins can bind viral surface proteins and destabilize them, effectively disabling the virus before it contacts a host cell. Kudryashova et al. (2022) demonstrated this mechanism against SARS-CoV-2, showing that HNP1 bound the spike protein with submicromolar affinity and promoted its unfolding and aggregation. The defensin locked the spike protein in a non-native, partially unfolded, nonfunctional state. This protein-destabilizing activity also extended to other viral glycoproteins, suggesting a general mechanism rather than one specific to any single virus.[2]
Receptor blocking. Some defensins prevent viral entry by binding host cell receptors rather than (or in addition to) the virus. Xu et al. (2021) showed that human defensin HD5 blocked SARS-CoV-2 infection by binding directly to ACE2, the host receptor the virus uses for cell entry. HD5 physically occupied the spike protein binding site on ACE2, preventing viral attachment. This receptor-cloaking mechanism is distinct from the direct viral neutralization seen with HNP1 and provides a second, independent barrier to infection.[3]
Fusion inhibition. After a virus binds its receptor, it must fuse its membrane with the host cell membrane (for enveloped viruses) or undergo conformational changes to inject its genome (for non-enveloped viruses). Defensins can interfere with both processes. Kudryashova et al. showed that HNP1 and retrocyclin RC-101 both inhibited spike-mediated membrane fusion, blocking a step downstream of receptor binding.[2] Skeate et al. (2020) demonstrated that theta-defensins inhibited high-risk human papillomavirus (HPV) infection through a charge-driven capsid clustering mechanism that prevented viral uncoating.[12]
Immune modulation. Beyond their direct antimicrobial effects, defensins serve as immunomodulatory molecules that bridge innate and adaptive immunity. Kim et al. (2018) demonstrated that HBD-2 potentiated innate antiviral immunity by enhancing interferon-stimulated gene (ISG) expression and promoting antigen-specific adaptive immune responses. The immunomodulatory effects of HBD-2 extended beyond its direct antiviral activity, amplifying the broader immune response to viral challenge.[4]
These four mechanisms are not mutually exclusive. A single defensin may simultaneously destabilize viral proteins, block receptors, inhibit fusion, and activate immune cells. This multi-pronged attack explains why defensins retain antiviral activity against diverse virus families, including both enveloped (influenza, HIV, SARS-CoV-2) and non-enveloped (adenovirus, HPV, rotavirus) viruses.
Defensins versus SARS-CoV-2: pandemic-era evidence
The COVID-19 pandemic generated a wave of research on defensin-virus interactions, producing both mechanistic insights and clinical correlations.
Xu et al. (2021) tested a panel of human defensins against SARS-CoV-2 pseudoviruses expressing the spike protein. HNP1, HNP2, HNP3, HD5, HBD-2, and HBD-3 all inhibited pseudovirus entry, though potencies varied. HD5 was particularly notable because it blocked infection through the receptor-cloaking mechanism described above, physically preventing spike from engaging ACE2.[3]
Kudryashova et al. (2022) extended these findings using authentic SARS-CoV-2 virus. HNP1 and retrocyclin RC-101 both blocked authentic viral infection in cell culture. The defensins were effective against not just the original strain but also the B.1.1.7 (Alpha) and P.1 (Gamma) variants, which carried mutations in the spike protein's receptor-binding domain. This broad-variant activity is consistent with the protein-destabilizing mechanism: rather than targeting a specific epitope that mutations could alter, the defensins promoted global structural disruption of the spike trimer.[2]
However, serum proteins diminished the anti-SARS-CoV-2 activity of HNP1 and RC-101. This finding has practical implications: defensins may be most effective as topical antiviral agents (intranasal sprays, for example) rather than systemic therapeutics, because serum albumin and other blood proteins compete for defensin binding and reduce the effective concentration available to neutralize virus.
Qian et al. (2023) provided clinical correlation data from a multi-omic analysis of COVID-19 patients. Plasma levels of alpha-defensin-1 (DEFA1/HNP1) correlated with disease severity: higher defensin levels were found in patients with more severe disease. This correlation likely reflects greater neutrophil activation and degranulation in severe cases rather than a causal role for defensins in pathology. Importantly, the researchers confirmed that DEFA1 inhibited SARS-CoV-2 pseudovirus infection in vitro, supporting the interpretation that elevated defensin levels represent an active, though ultimately insufficient, immune response.[9]
Defensins and influenza: the airway first line
Four Mechanism Categories
How Antiviral Peptides Actually Stop Influenza
The most promising therapeutic approach combines at least two of these. A peptide that both physically wrecks virions and boosts interferon would handle both ongoing and future infections. Most current single-peptide candidates do only one.
Source: Agamennone et al. (2022); Tripathi et al. (2013); Tongaonkar et al. (2025)
 View as image
View as imageThe interplay between defensins and influenza viruses has been studied for over two decades.
Human beta-defensins are expressed in the respiratory epithelium where influenza viruses first make contact. HBD-1 is constitutively present on the airway surface, providing a baseline defense. Upon influenza infection, HBD-2 expression is rapidly upregulated in airway epithelial cells both in vitro and in vivo. Kim et al. (2018) showed that HBD-2 did not just directly inhibit influenza. It enhanced the innate antiviral response by promoting interferon signaling, creating a feed-forward loop where defensin release amplified the broader antiviral state of the tissue.[4]
Alpha-defensins contribute through a different pathway. When neutrophils are recruited to influenza-infected airways, they release HNP1-3 in large quantities. Tripathi et al. (2013) compared the anti-influenza mechanisms of LL-37 (the human cathelicidin, covered in cathelicidins and respiratory viruses) and human defensins, finding that the two peptide families inhibit influenza through distinct mechanisms. While LL-37 directly disrupted viral membranes, defensins primarily aggregated viral particles, reducing the number of infectious virions able to bind and enter epithelial cells.[11]
This mechanistic complementarity between defensins and cathelicidins in the airway is significant. The two peptide families are co-expressed in respiratory tissue, and their distinct anti-influenza mechanisms mean they provide additive rather than redundant protection. A virus that evolved resistance to one mechanism would still face the other. For a broader overview of peptide defenses against influenza, including non-defensin peptides, see antiviral peptides against influenza.
The temporal dynamics of defensin expression during influenza infection follow a specific pattern. HBD-1 is already present on the airway surface before the virus arrives, serving as a constitutive sentine. Within hours of influenza infection, epithelial cells upregulate HBD-2 through NF-kB and toll-like receptor signaling, increasing local defensin concentrations severalfold. Simultaneously, the inflammatory response recruits neutrophils that degranulate and release stored alpha-defensins into the airway fluid. This layered response, constitutive baseline defense followed by induced epithelial production followed by neutrophil-delivered reinforcement, creates escalating defensin concentrations that track with viral replication kinetics.
The concentration dependence matters because defensin antiviral activity is typically dose-responsive. Below threshold concentrations, defensins may slow but not halt viral spread. Above threshold concentrations, particularly in the concentrated environment of airway surface liquid (which forms a thin layer just micrometers thick over the epithelium), defensins can achieve the local peptide densities needed for effective viral neutralization. This biophysical reality explains why the airway surface, where defensins accumulate in a confined fluid layer, may be the environment most conducive to defensin-mediated antiviral defense.
Genetic Variation
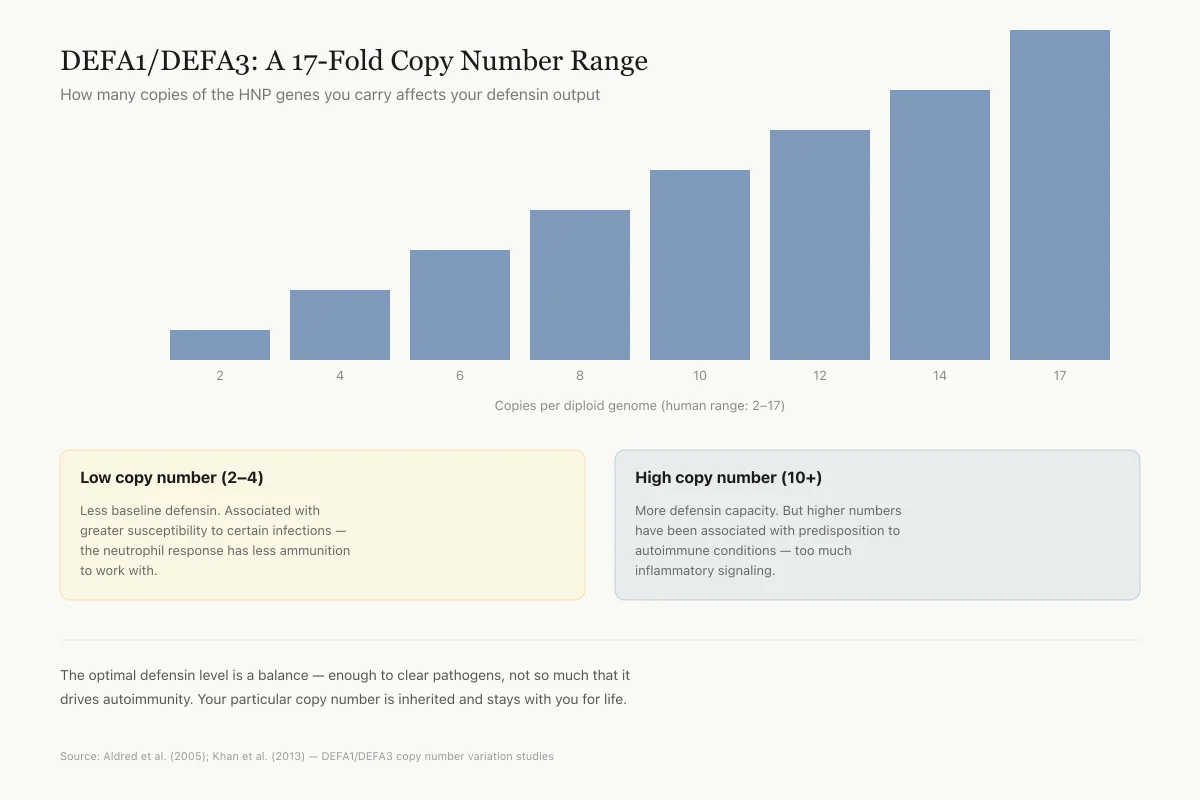
DEFA1/DEFA3: A 17-Fold Copy Number Range
How many copies of the HNP genes you carry affects your defensin output
Copies per diploid genome (human range: 2–17)
Low copy number (2–4)
Less baseline defensin. Associated with greater susceptibility to certain infections — the neutrophil response has less ammunition to work with.
High copy number (10+)
More defensin capacity. But higher numbers have been associated with predisposition to autoimmune conditions — too much inflammatory signaling.
This is one of the most dramatic examples of structural variation in the human genome. The optimal defensin level is a balance — enough to clear pathogens, not so much that it drives autoimmunity. Your particular copy number is inherited and stays with you for life.
Source: Aldred et al. (2005); Khan et al. (2013) — DEFA1/DEFA3 copy number variation studies
 View as image
View as imageViral immune evasion: when pathogens fight back
Defensins are not a static barrier. Viruses evolve to evade them.
Idris et al. (2022) found that SARS-CoV-2 infection led to downregulation of defensin gene expression in COVID-19 patients. Reduced HBD-1, HBD-4, and HD5 transcript levels were observed in infected individuals compared to healthy controls, suggesting that the virus actively suppresses the innate peptide defense system. This immunosuppressive strategy would create a more permissive environment for viral replication in the airway epithelium.[10]
Gilbert et al. (2021) added an age dimension to this picture. Analyzing nasopharyngeal samples from SARS-CoV-2-infected individuals of different ages, they found that beta-defensin expression varied with age, potentially contributing to the well-documented age-related differences in COVID-19 severity. Older patients, who are most vulnerable to severe disease, showed distinct patterns of interferon and defensin expression compared to younger patients.[5]
Diaz and Smith (2020) described a more fundamental evasion strategy in PLoS Pathogens. Their study demonstrated that defensins exert measurable evolutionary pressure on viral populations. When adenovirus populations were passaged in the presence of alpha-defensins, the virus evolved reduced sensitivity to defensin-mediated neutralization within relatively few generations. This "defensin-driven viral evolution" represents an arms race between host innate immunity and viral adaptation, analogous to the well-described co-evolution between viruses and adaptive immune responses.[6]
This evolutionary dimension has implications for therapeutic development. Any strategy that relies on defensins or defensin-derived molecules as antivirals must account for the possibility that sustained viral exposure to defensin pressure could select for resistant variants, similar to antibiotic resistance in bacteria.
The arms race metaphor is apt but should not be overstated. Defensin resistance in viruses requires changes to fundamental structural properties (envelope lipid composition, surface protein stability, receptor-binding domain architecture) that carry substantial fitness costs. Bacteria that evolve resistance to antimicrobial peptides often suffer growth rate penalties or reduced virulence. Whether viruses face analogous constraints on defensin resistance evolution is not well characterized, but the multi-mechanism nature of defensin activity (targeting both viral structure and host immune pathways simultaneously) creates a higher evolutionary barrier than single-target antiviral drugs.
The relationship between defensin exposure and viral fitness is further complicated by the fact that defensin concentrations fluctuate dynamically during infection. A virus that encounters maximal defensin pressure during acute inflammation may face very different selective conditions during chronic or latent infection when defensin levels are lower. This heterogeneous selection landscape may prevent complete resistance evolution by maintaining viral subpopulations sensitive to defensin neutralization.
Therapeutic potential and limitations
Solanki et al. (2021) reviewed the prospects for defensins as antiviral therapeutics, noting both promise and obstacles. The direct antiviral activity, broad-spectrum efficacy across virus families, and immunomodulatory properties make defensins attractive candidates. Unlike conventional antivirals that target specific viral enzymes, defensins attack fundamental features of viral architecture (envelope lipids, surface protein stability, receptor interactions) that are difficult for viruses to alter without losing fitness.[7]
Zupin et al. (2022) expanded this perspective, arguing that defensins could serve dual roles as both direct antivirals and vaccine adjuvants. Their immunomodulatory properties, particularly the ability of beta-defensins to enhance antigen-specific adaptive immune responses, could improve vaccine efficacy when defensins are co-administered with viral antigens.[8]
Tongaonkar et al. (2025) demonstrated that rhesus theta-defensin 1 (RTD-1) activated interferon and antiviral pathways in human monocytes, providing mechanistic evidence for the immunomodulatory activity of cyclic defensins. RTD-1 induced expression of type I interferons and interferon-stimulated genes, suggesting that theta-defensin analogs could serve as immune activators for prophylactic or therapeutic use against respiratory viruses.[13]
The limitations are substantial. Serum binding reduces defensin potency systemically. Manufacturing cysteine-rich disulfide-bonded peptides at pharmaceutical scale is expensive. The narrow window between antiviral and cytotoxic concentrations for some defensins limits dose escalation. And the evolutionary evidence from Diaz and Smith suggests that long-term therapeutic use could drive viral resistance.
The most realistic near-term applications may be topical: intranasal defensin formulations for prophylaxis against respiratory viruses, or defensin-based wound treatments for herpes simplex or HPV lesions where the peptides can be delivered directly to the site of infection at effective concentrations without systemic exposure.
The adjuvant approach may prove more immediately tractable than direct antiviral use. Defensins naturally bridge innate and adaptive immunity, and several research groups have explored incorporating defensin sequences or defensin-derived motifs into vaccine formulations to enhance immunogenicity. If successful, this strategy would leverage the immunomodulatory properties of defensins without requiring the high local concentrations needed for direct viral neutralization, potentially sidestepping the therapeutic window and serum binding challenges that complicate antiviral applications.
The antimicrobial peptide field more broadly faces similar translational challenges. Defensins share the class-wide issues of manufacturing cost, stability in biological fluids, and the need for careful selectivity optimization to avoid host cell toxicity. Lessons from other antimicrobial peptides in clinical development, including the importance of formulation science, delivery route optimization, and combination strategies, apply directly to defensin-based antiviral candidates.
What the evidence does not yet show
Several gaps remain in the defensin-respiratory virus evidence base.
No clinical trial has tested defensin-based antivirals in human respiratory infections. All anti-influenza and anti-SARS-CoV-2 data come from cell culture systems, animal models, or observational clinical correlations. The step from laboratory efficacy to clinical benefit has not been taken.
The relationship between natural defensin levels and infection outcomes is correlational, not causal. Higher defensin levels in severe COVID-19 may reflect neutrophil activation during inflammation rather than indicating that defensins contribute to disease severity. Conversely, lower defensin gene expression in infected patients could be a consequence of infection rather than a cause of susceptibility.
The interaction between defensins and the respiratory microbiome is poorly characterized. Defensins that kill respiratory viruses could also alter commensal bacterial populations in the airway, with unknown consequences for mucosal immunity and disease susceptibility.
Whether synthetic retrocyclins (theta-defensins) offer practical advantages over natural alpha and beta-defensins as therapeutic candidates remains to be determined. Their cyclic structure provides greater stability, but manufacturing and safety data in humans are lacking.
The dose-response relationship for defensin antiviral activity in vivo has not been established for any respiratory virus. Cell culture experiments use defined peptide concentrations, but the actual defensin concentrations achieved in airway surface liquid during natural infection are difficult to measure and likely vary substantially between individuals, across different regions of the respiratory tract, and over the course of infection. Without this pharmacokinetic understanding, it is impossible to determine whether natural defensin levels are sufficient for meaningful antiviral protection or whether therapeutic supplementation would need to achieve supra-physiological concentrations to confer benefit.
Finally, the interaction between defensins and the mucus layer that lines the airways adds an additional layer of complexity. Mucins can bind and sequester antimicrobial peptides, potentially reducing the effective defensin concentration at the epithelial surface where viral entry occurs. Whether mucus acts as a reservoir that concentrates defensins at the mucosal surface or as a sink that dilutes them is not resolved, and the answer may differ between healthy individuals and those with mucus hypersecretion from conditions like chronic obstructive pulmonary disease or cystic fibrosis.
The Bottom Line
Human defensins provide multi-layered antiviral defense in the airways through direct viral disruption, receptor blocking, fusion inhibition, and immune activation. Evidence from the COVID-19 pandemic confirmed that multiple defensin classes can neutralize SARS-CoV-2, with HNP1 destabilizing the spike protein and HD5 cloaking the ACE2 receptor. Against influenza, defensins aggregate viral particles while beta-defensins amplify interferon signaling. Viruses fight back by downregulating defensin expression and evolving reduced sensitivity. Therapeutic applications face challenges from serum binding, narrow therapeutic windows, and manufacturing costs, but topical defensin formulations for respiratory virus prophylaxis remain a realistic near-term goal.
Sources & References
- 1RPEP-02303·Wilson, Sarah S et al. (2013). “Antiviral mechanisms of human defensins..” Journal of molecular biology.Study breakdown →PubMed →↩
- 2RPEP-06272·Kudryashova, Elena et al. (2022). “Human Defensin Peptides Can Block SARS-CoV-2 Infection by Destabilizing the Spike Protein.” Journal of molecular biology.Study breakdown →PubMed →↩
- 3RPEP-05890·Xu, Chuan et al. (2021). “Human Defensins Inhibit SARS-CoV-2 Infection by Blocking Viral Entry..” Viruses.Study breakdown →PubMed →↩
- 4RPEP-03752·Kim, Ju et al. (2018). “Human β-defensin 2 plays a regulatory role in innate antiviral immunity and is capable of potentiating the induction of antigen-specific immunity..” Virology journal.Study breakdown →PubMed →↩
- 5RPEP-05405·Gilbert, Charly et al. (2021). “Children and Adults Mount Different Defensin and Interferon Responses to COVID-19.” Frontiers in immunology.Study breakdown →PubMed →↩
- 6RPEP-04773·Diaz, Karina et al. (2020). “How Defensin Peptides Drive Virus Evolution: Adenovirus Mutates to Escape Gut Immune Defense.” PLoS pathogens.Study breakdown →PubMed →↩
- 7RPEP-05778·Solanki, Subhash Singh et al. (2021). “Promising role of defensins peptides as therapeutics to combat against viral infection..” Microbial pathogenesis.Study breakdown →PubMed →↩
- 8RPEP-06660·Zupin, Luisa et al. (2022). “Human Defensins from Antivirals to Vaccine Adjuvants: Rediscovery of the Innate Immunity Arsenal..” Protein and peptide letters.Study breakdown →PubMed →↩
- 9RPEP-07296·Qian, Xijing et al. (2023). “Multi-omic and comparative analyses revealed monocyte-derived alpha-defensin-1 correlated with COVID-19 severity and inhibited SARS-CoV-2 infection..” Journal of medical virology.Study breakdown →PubMed →↩
- 10RPEP-06217·Idris, Mohammed M et al. (2022). “Key Defensin Peptide Genes Are Suppressed During COVID-19 Infection.” Acta virologica.Study breakdown →PubMed →↩
- 11RPEP-02295·Tripathi, Shweta et al. (2013). “The human cathelicidin LL-37 inhibits influenza A viruses through a mechanism distinct from that of surfactant protein D or defensins..” The Journal of general virology.Study breakdown →PubMed →↩
- 12RPEP-05142·Skeate, Joseph G et al. (2020). “Monkey-Derived Defensin Peptides Block HPV Infection by Clumping Virus Particles Together.” Frontiers in immunology.Study breakdown →PubMed →↩
- 13RPEP-13821·Tongaonkar, Prasad et al. (2025). “Cyclic defensin peptide RTD-1 activates interferon antiviral pathways and blocks SARS-CoV-2 infection in human cells.” Journal of leukocyte biology.Study breakdown →PubMed →↩