Deep Learning for Peptide Property Prediction
AI in Peptide Science
49+ ML models
Over 49 studies in the RethinkPeptides database apply machine learning or deep learning to peptide property prediction, with the majority published since 2024.
RethinkPeptides Research Database, 2026
RethinkPeptides Research Database, 2026
If you only read one thing
Every peptide drug candidate has to pass a list of tests: does it hit the target, does it spare healthy cells, does it survive in blood, can it cross a membrane? Testing all of that by hand in a lab takes months per peptide. Deep learning models now read an amino acid sequence and predict most of those answers in seconds, which lets scientists pick which 50 of a million possible peptides to actually make. The predictions are useful, not magic — about 30 to 70 percent of the ones a good model picks as winners really do work in the lab, which is wildly better than random but still far from perfect.
Peptide drug development has historically been slow and expensive. Identifying a peptide with the right combination of properties (high target activity, low toxicity, cell permeability, protease stability, no hemolytic activity) required synthesizing and testing thousands of candidates. Each property was measured in separate assays, each assay consumed material and time, and the search space of possible peptide sequences grows exponentially with length. A 20-amino-acid peptide has 20 to the power of 20 (over 10 to the power of 25) possible sequences. No laboratory can screen that space experimentally.
Deep learning changed the equation. Neural networks trained on existing peptide datasets can now predict multiple properties directly from amino acid sequences, prioritizing candidates computationally before synthesis. Transformer architectures adapted from natural language processing treat peptide sequences as "languages" with grammar rules that encode biological function. Generative models design novel sequences with specified property profiles. The result is a fundamental acceleration of the design-make-test cycle that defines peptide drug discovery. What took months of synthesis and screening can now be narrowed to days of computation followed by targeted experimental validation of the most promising candidates. The field is moving fast: the majority of published peptide prediction models were released after 2023, and new architectures appear monthly.
Key Takeaways
- Deep learning models predict antimicrobial activity, toxicity, cell penetration, hemolytic potential, and structural properties from peptide amino acid sequences alone
- Transformer-based architectures (including ESM protein language models) have become the dominant approach, treating sequences as biological languages[1]
- PeptideNet integrates multiple deep learning modules to predict diverse bioactive peptide classes simultaneously rather than requiring separate models[2]
- Generative deep learning models now design novel antimicrobial peptides de novo, with experimentally validated activity against resistant bacteria[3]
- AlphaFold2 has been adapted for cyclic peptide structure prediction and stabilizer design, bridging structure prediction and drug design[4]
- Explainable AI methods are emerging alongside prediction accuracy, enabling researchers to understand why a model predicts a peptide will be active or toxic[5]
What "Property Prediction" Means for Peptides
A peptide's biological behavior is determined by its amino acid sequence. That sequence dictates the peptide's three-dimensional structure, which in turn determines how it interacts with biological targets: cell membranes, receptors, enzymes, and other proteins. Property prediction is the task of computationally inferring these behaviors from the sequence alone, without synthesizing the peptide or running experiments.
The properties most commonly predicted by deep learning models include:
Antimicrobial activity. Will this peptide kill or inhibit bacteria? At what minimum inhibitory concentration? Against which bacterial species? This is the most actively researched prediction task, driven by the urgent need for new antibiotics. For background on antimicrobial peptides, see Antimicrobial Peptides as Alternatives to Antibiotics: Can They Solve Resistance?. For the ML-specific story, see Machine Learning for Antimicrobial Peptide Prediction: Finding New Antibiotics.
Toxicity and hemolytic activity. Will this peptide damage human cells? Hemolysis (destruction of red blood cells) is the most common toxicity concern for antimicrobial peptides, because the same membrane-disrupting activity that kills bacteria can also damage human cell membranes.
Cell-penetrating ability. Can this peptide cross cell membranes to reach intracellular targets? Cell-penetrating peptides (CPPs) are critical for drug delivery, and predicting which sequences have this property accelerates development of intracellular therapeutics.
Structure and stability. What three-dimensional conformation will this peptide adopt? How stable is that conformation under physiological conditions? Structure prediction intersects with property prediction because many biological activities depend on specific structural features. See AlphaFold and Peptide Structure: Predicting Shape from Sequence.
Target binding. Will this peptide bind to a specific protein, receptor, or enzyme? With what affinity? This is the most complex prediction task and typically requires structural information about both the peptide and its target. AlphaFold Multimer and related structure prediction tools have improved binding prediction by modeling the three-dimensional interface between peptide and target protein, but accurate binding affinity prediction (predicting Kd values) remains one of the field's hardest unsolved problems.
Stability and half-life. How long will this peptide survive in serum, gastric fluid, or intracellular environments? Protease susceptibility depends on local sequence context and three-dimensional accessibility of cleavage sites. Models trained on experimentally measured stability data can identify sequences likely to resist degradation, guiding the design of peptides with improved pharmacokinetic properties.
How Deep Learning Approaches Work
Architecture generations
From Decision Trees to Protein Language Models
Every generation unlocked something its predecessor could not see.
2015–2018
Classical ML (SVM, random forest, gradient boosting)
Good at: Works with tiny datasets, fast to train
Still hard: Needs handcrafted features; poor for quantitative prediction
2018–2020
Convolutional networks (CNNs)
Good at: Picks up local sequence motifs automatically
Still hard: Misses long-range relationships across the sequence
2019–2021
Recurrent networks (RNN, LSTM)
Good at: Processes sequences position by position with memory
Still hard: Struggles on longer peptides; training instability
2020–present
Transformers + protein language models (ESM)
Good at: Captures every-position-to-every-position relationships; benefits from pretraining on millions of proteins
Still hard: Data-hungry; opaque unless paired with explainability tools
2023–present
Graph neural networks + multimodal ensembles
Good at: Adds 3-D structural context to sequence features
Still hard: Complex to train; reproducibility is uneven
Source: Lin et al. (2026); field review across peptide ML literature
 View as image
View as imageFrom Sequence to Feature Representation
The first challenge in applying deep learning to peptides is converting an amino acid sequence into a numerical representation that neural networks can process. Several approaches exist:
One-hot encoding represents each amino acid as a 20-dimensional binary vector. Simple but loses information about amino acid similarity (leucine and isoleucine are treated as equally different as leucine and glutamate).
Physicochemical descriptors encode each amino acid by properties like hydrophobicity, charge, molecular weight, and hydrogen bonding capacity. This captures chemical similarity but requires choosing which properties matter.
Protein language model embeddings use pretrained transformer models (ESM, ProtBert, ProtTrans) that have learned distributed representations of amino acids by training on millions of protein sequences. These embeddings capture evolutionary and structural relationships automatically, without manual feature engineering. ESM (Evolutionary Scale Modeling) embeddings have become the dominant input representation for state-of-the-art peptide prediction models.
Architecture Evolution
The field has progressed through several architectural generations:
Traditional machine learning (random forests, SVMs, gradient boosting) dominated early work (2015-2018). These models used handcrafted features and achieved reasonable accuracy for binary classification (antimicrobial vs. non-antimicrobial) but struggled with quantitative predictions and generalization.
Convolutional neural networks (CNNs) treated peptide sequences as 1D signals and identified local sequence motifs associated with activity. They improved on traditional ML for pattern recognition but could not capture long-range sequence dependencies.
Recurrent neural networks (RNNs/LSTMs) processed sequences position by position, maintaining memory of earlier residues. Better at capturing sequence-level patterns but limited by vanishing gradient problems in longer sequences.
Transformers and attention mechanisms (2020-present) process entire sequences simultaneously using self-attention, identifying which amino acid positions influence each other regardless of distance in the sequence. This architecture, borrowed from natural language processing, has produced the largest accuracy gains in peptide property prediction. PepGraphormer, for example, combines ESM protein language model features with graph attention networks to predict antimicrobial activity, achieving state-of-the-art performance by representing peptides as heterogeneous graphs that capture both sequential and structural relationships.[1]
Graph Neural Networks
A newer approach represents peptides not as linear sequences but as molecular graphs, where atoms or amino acids are nodes and chemical bonds or spatial contacts are edges. Graph neural networks (GNNs) process these graph representations to capture three-dimensional structural relationships that linear sequence models miss. PepGraphormer combines graph attention with ESM embeddings, leveraging both sequential and structural information.[1]
Ensemble and Multi-Modal Approaches
State-of-the-art prediction systems increasingly combine multiple model types. An ensemble might use a transformer for sequence-level features, a CNN for local motif detection, and a GNN for structural features, then aggregate their predictions through a meta-learner. This multi-modal approach reduces the risk of any single architecture's blind spots and typically outperforms individual models on benchmark datasets.
Key Models and Platforms
The current model lineup
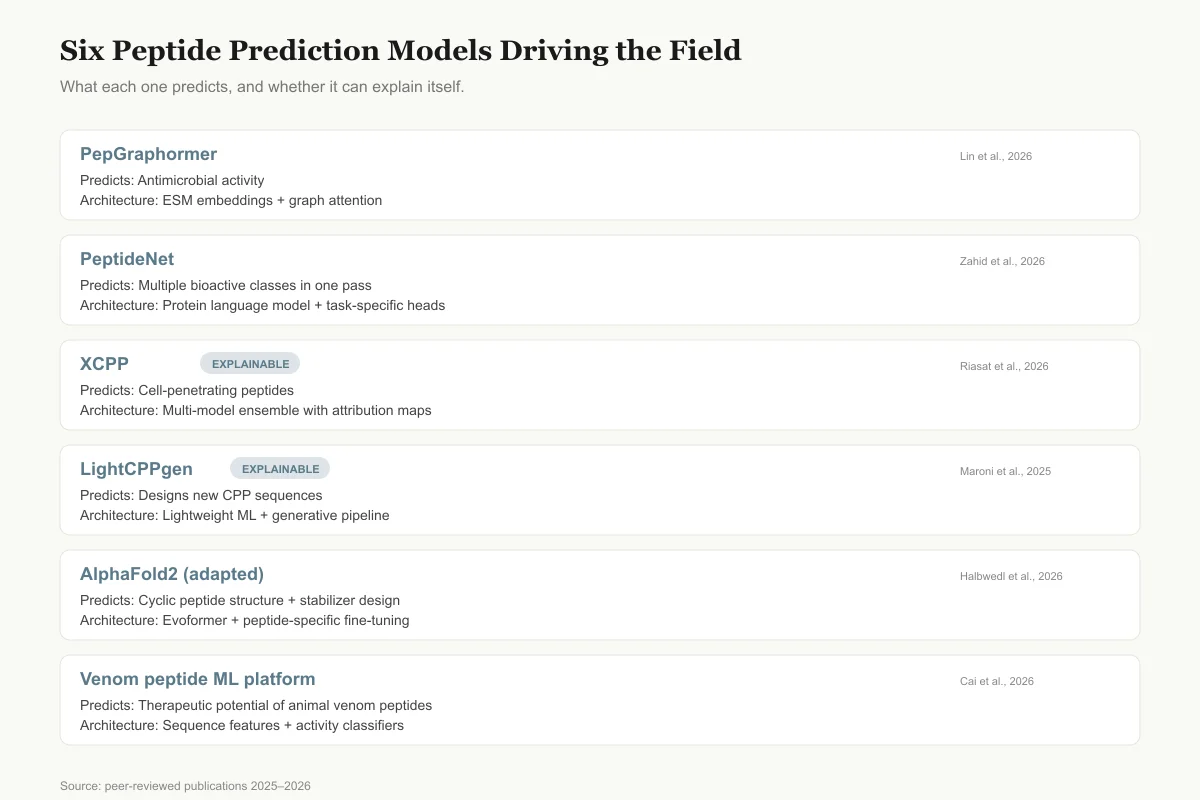
Six Models Driving Peptide Prediction in 2025–2026
What each one actually predicts, and whether it can explain itself.
PepGraphormer
Predicts
Antimicrobial activity
Architecture
ESM embeddings + graph attention network
Lin et al., 2026
PeptideNet
Predicts
Multiple bioactive classes in one pass
Architecture
Protein language model + task-specific heads
Zahid et al., 2026
XCPP
ExplainablePredicts
Cell-penetrating peptides
Architecture
Multi-model ensemble with attribution maps
Riasat et al., 2026
LightCPPgen
ExplainablePredicts
Designs new CPP sequences
Architecture
Lightweight ML + generative pipeline
Maroni et al., 2025
AlphaFold2 (adapted)
Predicts
Cyclic peptide structure + stabilizer design
Architecture
Evoformer + peptide-specific fine-tuning
Halbwedl et al., 2026
Venom peptide ML platform
Predicts
Therapeutic potential of animal venom peptides
Architecture
Sequence features + activity classifiers
Cai et al., 2026
Source: Peer-reviewed publications 2025–2026 (see article references)
 View as image
View as imagePeptideNet: Multi-Property Prediction
Most early models predicted a single property. PeptideNet, published in 2026, introduced an integrative deep learning framework that predicts diverse bioactive peptide classes using protein language model features. Rather than training separate models for antimicrobial, antioxidant, anti-inflammatory, and other activities, PeptideNet uses a shared representation with task-specific output heads, allowing it to predict multiple properties simultaneously from a single input sequence.[2]
XCPP: Cell-Penetrating Peptide Prediction
Riasat and colleagues developed XCPP, a multi-model explainable deep learning framework for identifying cell-penetrating peptides. The "explainable" aspect is critical: XCPP not only predicts whether a peptide will penetrate cells but highlights which sequence features drive the prediction, enabling researchers to rationally modify sequences to enhance or reduce penetration.[5]
LightCPPgen: Explainable Design
Maroni and colleagues took explainability further with LightCPPgen, a machine learning pipeline for rational design of cell-penetrating peptides. The model identifies sequence patterns associated with cell penetration and uses them to generate novel CPP sequences with predicted activity, combining prediction with generative design.[6]
AlphaFold2 for Cyclic Peptide Design
DeepMind's AlphaFold2 was originally developed for protein structure prediction, but researchers have adapted it for peptide applications. Halbwedl and colleagues used AlphaFold2 to guide cyclic peptide stabilizer design targeting protein-protein interactions, demonstrating that the model's structural predictions are accurate enough to inform medicinal chemistry decisions for cyclic peptide drug candidates.[4]
Venom Peptide Discovery
Cai and colleagues reported a machine learning-enabled platform for rapid drug discovery from animal venom peptides. Venoms contain thousands of bioactive peptides evolved over millions of years, but characterizing them experimentally is slow. The ML platform predicts which venom peptide sequences are likely to have therapeutic activity, dramatically narrowing the candidates for synthesis and testing.[7]
Generative Models: Designing New Peptides
Prediction models evaluate existing sequences. Generative models create new ones. This is the frontier of AI-driven peptide science.
Liu and colleagues demonstrated de novo design of self-assembling antimicrobial peptides guided by deep learning. The generative model produced novel sequences not found in any database, and experimental testing confirmed antimicrobial activity against resistant bacteria.[3] This represents a fundamental shift: rather than mining nature's peptide library, researchers can now computationally design peptides that never existed in any organism.
Mechesso and colleagues reported a one-step design approach for potent, nonhemolytic antimicrobial peptides using a database-guided, non-machine-learning optimization. While not deep learning per se, this work illustrates the complementary role of computational design in creating peptides that achieve the difficult combination of high antimicrobial potency with low human cell toxicity.[8]
For a comprehensive overview of how generative AI is changing peptide design, see Generative AI for Novel Peptide Design: Creating Molecules That Never Existed. For the broader impact on the drug discovery pipeline, see How AI Is Revolutionizing Peptide Drug Discovery.
Understanding Mechanisms: Beyond Black Boxes
A 2026 study by Bouvier and colleagues applied transformer architectures not to predict activity but to understand mechanism. By classifying antimicrobial peptide conformational dynamics using molecular dynamics simulation data as input, they demonstrated that transformers can identify the physical motions that distinguish active from inactive peptides. This "motion as a language" approach bridges the gap between sequence-based prediction and physics-based understanding.[9]
Malshikare and colleagues took a different approach to mechanistic understanding, using charge density-based machine learning to uncover principles of antimicrobial peptide action. Rather than treating the model as a black box, they extracted interpretable features that revealed how charge distribution along the peptide sequence determines membrane interaction and bacterial killing.[10]
Cysteine pattern barcoding, developed by Bibi and colleagues, uses the arrangement of cysteine residues (which form disulfide bonds) as a classification system for peptide families, enabling machine learning models to leverage structural information encoded in the sequence without requiring explicit structure prediction.[11]
These mechanistic approaches represent a maturation of the field from pure prediction (does this peptide work?) to understanding (why does it work?). Understanding why enables rational optimization in ways that pure prediction cannot.
Real-World Impact: From Models to Molecules
The practical impact of deep learning on peptide science can be measured by how many computationally designed peptides have been experimentally validated. The answer is growing but still modest relative to the volume of computational predictions published.
The most convincing examples come from antimicrobial peptide design. Multiple groups have used deep learning to generate novel AMP sequences, synthesized them, and demonstrated antimicrobial activity against drug-resistant bacteria in vitro. Liu and colleagues' self-assembling AMPs are one example.[3] The hit rates from these studies (fraction of predicted-active sequences that test positive) are typically 30-70% for well-constrained design problems, compared to less than 1% for random sequence screening.
In drug discovery pipelines, deep learning is primarily used at the hit identification and lead optimization stages. The computational models narrow millions of possible sequences to hundreds of candidates, which are then synthesized and tested in standard biological assays. This acceleration is most impactful in the early stages of discovery, where the search space is largest and experimental resources are most constrained.
The field has also begun to produce open-source tools that non-specialists can use. Web servers for AMP prediction (CAMP, ADAM, iAMPpred) and downloadable models hosted on GitHub have democratized access to peptide property prediction, though interpreting and acting on predictions still requires domain expertise.
One area where computational prediction has had less impact than expected is clinical translation. Despite hundreds of published prediction models and thousands of computationally designed peptides, no AI-designed peptide has yet completed clinical trials and reached market. The bottleneck is not the computational design but the downstream development process: formulation, pharmacokinetics, toxicology, and manufacturing at scale remain largely experimental rather than computational challenges.
Limitations and Challenges
Training-data gap
Peptide Databases Are Tiny by Deep-Learning Standards
Bars scaled within the chart. The actual gap between peptide datasets and general LLM training data spans seven orders of magnitude.
APD3 (Antimicrobial Peptide Database)
~3,000 peptides
DBAASP (peptide activity + structure)
~18,000 peptides
DRAMP (antimicrobial repository)
~20,000 peptides
UniRef50 (protein sequences used to pretrain ESM)
~60,000,000 sequences
Common Crawl (used to train large language models)
Hundreds of billions of tokens
Why it matters: peptide prediction accuracy is limited less by algorithms and more by the fact that activity-labeled peptide data is scarce, biased toward positives, and often irreproducible across labs.
Source: APD3; DBAASP; DRAMP; UniRef50; public LLM training-set disclosures
 View as image
View as imageTraining data quality and bias. Deep learning models are only as good as their training data. Peptide activity databases contain experimental measurements from different laboratories using different protocols, creating noise and inconsistency. The Antimicrobial Peptide Database (APD3) contains approximately 3,000 entries. The Database of Antimicrobial Activity and Structure of Peptides (DBAASP) contains over 18,000 entries. The DRAMP database contains over 20,000 entries. These databases are large by peptide standards but small by deep learning standards, where language models train on billions of tokens. Most antimicrobial peptide databases are heavily biased toward peptides that were tested because they were expected to be active, underrepresenting the negative space of inactive sequences. This positive bias inflates apparent model accuracy and reduces real-world predictive reliability.
Generalization across peptide classes. A model trained on alpha-helical antimicrobial peptides may perform poorly on beta-sheet antimicrobial peptides or cyclic peptides. Domain adaptation and transfer learning partially address this, but most models still perform best within the peptide class they were trained on.
Experimental validation gap. Publication bias favors computational studies with promising predictions. The rate of experimental validation of computationally designed peptides remains low. When validation is performed, the hit rate (fraction of predicted-active sequences that are actually active in assays) varies widely, from over 50% for well-constrained problems to under 10% for more ambitious designs.
Multi-objective optimization. A clinically useful peptide must be active against its target, non-toxic to human cells, stable in biological fluids, cell-permeable if needed, and manufacturable at scale. Optimizing all properties simultaneously is harder than optimizing any single property, and trade-offs between properties (e.g., membrane activity correlates with both antimicrobial potency and hemolytic toxicity) create fundamental design tensions that computational models must navigate. Pareto optimization and constrained generative models address this, but the multi-property landscape remains the hardest challenge in computational peptide design. Mechesso and colleagues' approach of designing potent yet nonhemolytic antimicrobial peptides illustrates the difficulty: achieving high antimicrobial potency without hemolysis required navigating a narrow region of sequence space.[8]
Interpretability. Most deep learning models operate as black boxes. XCPP and LightCPPgen represent efforts toward explainability, but the majority of published models cannot explain why they predict a given sequence to be active or inactive. This limits trust and adoption by medicinal chemists who need mechanistic rationale for design decisions.
Scalability of wet-lab validation. The computational capacity to screen millions of sequences exceeds the experimental capacity to validate them by orders of magnitude. This creates a bottleneck: models can generate thousands of promising candidates, but only dozens can be synthesized and tested in practice. Automated peptide synthesis platforms and high-throughput screening are partially closing this gap, but the mismatch between computational and experimental throughput remains the most practical limitation of the field. For the combinatorial approach to this challenge, see Combinatorial Peptide Libraries: The Shotgun Approach to Drug Discovery.
Reproducibility. Many published models are not accompanied by usable code, trained model weights, or sufficient documentation for independent researchers to reproduce results. Studies that do release code sometimes depend on specific software versions or hardware configurations that limit reusability. The field is improving through platforms like Hugging Face and standardized benchmarking datasets, but reproducibility remains inconsistent. Standardized benchmarking competitions, analogous to CASP for protein structure prediction, would accelerate progress by creating shared evaluation standards.
The Bottom Line
Deep learning has transformed peptide property prediction from a slow, experimental process into a computational one. Transformer architectures and protein language model embeddings now predict antimicrobial activity, toxicity, cell penetration, and structural properties from amino acid sequences with increasing accuracy. Generative models design novel peptides with experimentally validated activity. The field is moving toward multi-property prediction, explainability, and mechanistic understanding. The primary limitations are training data quality, the experimental validation gap, and the challenge of optimizing multiple properties simultaneously in a single peptide sequence.
Sources & References
- 1RPEP-15563·Lin, Changhang et al. (2026). “PepGraphormer: an ESM-GAT hybrid deep learning framework for antimicrobial peptide prediction..” Journal of cheminformatics.Study breakdown →PubMed →↩
- 2RPEP-16525·Zahid, Hamza et al. (2026). “PeptideNet: AI That Predicts Whether a Peptide Will Fight Viruses, Bacteria, or Oxidative Damage.” Journal of chemical information and modeling.Study breakdown →PubMed →↩
- 3RPEP-12210·Liu, Huayang et al. (2025). “De novo design of self-assembling peptides with antimicrobial activity guided by deep learning..” Nature materials.Study breakdown →PubMed →↩
- 4RPEP-15252·Halbwedl, Niklas et al. (2026). “AlphaFold2-Based Method Designs Cyclic Peptides That Stabilize Protein-Protein Interactions.” Proteins.Study breakdown →PubMed →↩
- 5RPEP-15995·Riasat, Hafsah et al. (2026). “XCPP: A Multi-model Explainable Deep Learning Framework for Accurate Identification of Cell-Penetrating Peptides from Structured Sequence Features..” Current drug targets.Study breakdown →PubMed →↩
- 6RPEP-12451·Maroni, Gabriele et al. (2025). “Machine Learning Pipeline Designs Cell-Penetrating Peptides While Explaining Why They Work.” International journal of antimicrobial agents.Study breakdown →PubMed →↩
- 7RPEP-14920·Cai, Fei et al. (2026). “Machine Learning Platform Mines Animal Venom Peptides for Rapid Drug Discovery.” Pharmaceuticals (Basel.Study breakdown →PubMed →↩
- 8RPEP-15696·Mechesso, Abraham F et al. (2026). “One-Step Design of Potent and Nonhemolytic Antimicrobial Peptides by Using a Database-Guided, Nonmachine Learning Approach..” ACS infectious diseases.Study breakdown →PubMed →↩
- 9RPEP-14901·Bouvier, Benjamin (2026). “Using Transformer AI to Classify Antimicrobial Peptides by How They Move.” Journal of chemical theory and computation.Study breakdown →PubMed →↩
- 10RPEP-15656·Malshikare, Hrushikesh et al. (2026). “Machine Learning Reveals Three Distinct Ways Antimicrobial Peptides Kill Bacteria Based on Charge.” Chemical communications (Cambridge.Study breakdown →PubMed →↩
- 11RPEP-10153·Bibi, Rimsha et al. (2025). “Cysteine pattern barcoding-based dataset filtration enhances the machine learning-assisted interpretation of Conus venom peptide therapeutics..” PloS one.Study breakdown →PubMed →↩