Combinatorial Peptide Libraries in Drug Discovery
Peptide Library Screening Technologies
10¹² unique sequences
mRNA display libraries, including the RaPID system, can generate and screen over one trillion unique macrocyclic peptide sequences against a single protein target.
Goto & Suga, Accounts of Chemical Research, 2021
Goto & Suga, Accounts of Chemical Research, 2021
If you only read one thing
A combinatorial peptide library is a laboratory trick for testing billions of different peptide drug candidates at the same time, in a single tube. Instead of designing one molecule and hoping it works, scientists generate massive random collections, expose them all to a target protein, and keep only the few that stick. Four platforms dominate: phage display (the Nobel-winning classic), OBOC (chemistry freedom), mRNA display (trillion-member screens), and DNA-encoded libraries (barcoded synthetic chemistry). Over 80 approved peptide drugs trace part of their lineage back to this idea.
Designing a peptide drug from scratch requires guessing which of the approximately 20^15 possible 15-residue sequences will bind a given target. Combinatorial peptide libraries eliminate the guessing. Instead of designing one candidate at a time, these technologies generate millions to trillions of different peptide sequences simultaneously, then use selection pressure to isolate the rare molecules that bind a target protein with high affinity. The approach is closer to evolution than engineering: create vast diversity, apply selective pressure, amplify the winners. Four major platform technologies now dominate the field, each with distinct advantages: phage display (10^10 diversity, genetically encoded), one-bead-one-compound (OBOC) libraries (10^6-10^8, chemically diverse), mRNA display (10^12-10^13, the largest libraries possible), and DNA-encoded libraries (DELs, 10^9-10^12, bridging chemistry and genetics). Together, these platforms have contributed to the discovery of peptide therapeutics generating over $50 billion in annual revenue.[10] This article maps each technology, its evidence base, and its role in modern peptide drug discovery. For technology-specific deep dives, see the dedicated articles on phage display, mRNA display, and high-throughput peptide screening.
Key Takeaways
- Phage display peptide libraries, first demonstrated by Scott and Smith in 1990, can screen approximately 10 billion unique peptide sequences against a protein target in a single experiment.[1]
- The OBOC method introduced by Lam et al. in 1991 displays one unique peptide per bead and allows incorporation of unnatural amino acids and cyclic structures that phage display cannot access.[3]
- The RaPID mRNA display system generates libraries exceeding 10^12 macrocyclic peptides and has produced binders with subnanomolar dissociation constants against targets considered undruggable by small molecules.[5]
- DNA-encoded macrocyclic peptide libraries of 2.4 trillion members have been screened against therapeutic targets, yielding selective inhibitors with IC50 values as low as 8.9 nM (Zhu et al., ACS Chemical Biology, 2018).[9]
- Phage display peptide libraries earned George Smith and Gregory Winter the 2018 Nobel Prize in Chemistry for enabling the directed evolution of peptides and antibodies.[4]
- Over 80 peptide drugs have reached clinical approval, with combinatorial library screening playing an increasingly central role in lead identification alongside rational design and natural product isolation.[10]
The core principle: diversity plus selection
All combinatorial peptide library approaches share a fundamental logic. First, generate an enormous collection of chemically distinct peptide sequences. Second, expose the entire collection to a molecular target, such as a receptor, enzyme, or cell surface. Third, separate the molecules that bind from those that do not. Fourth, identify the winning sequences. The power of the approach lies in the numbers: testing 10 billion candidates simultaneously is not 10 billion times more expensive than testing one. It is often only marginally more costly, because the entire library occupies a single test tube.
The concept draws directly from natural selection. Biological immune systems use a similar strategy: the adaptive immune system generates approximately 10^11 distinct antibody sequences through V(D)J recombination, then selects the rare clones that recognize a pathogen. Combinatorial peptide libraries are a laboratory distillation of this principle, compressed from weeks of immune response into hours of bench work.[4]
The technologies differ in how they create diversity, maintain the link between a peptide's structure and its identity (the genotype-phenotype link), and what chemical space they can access. These differences determine which platform is best suited for a given drug discovery campaign.
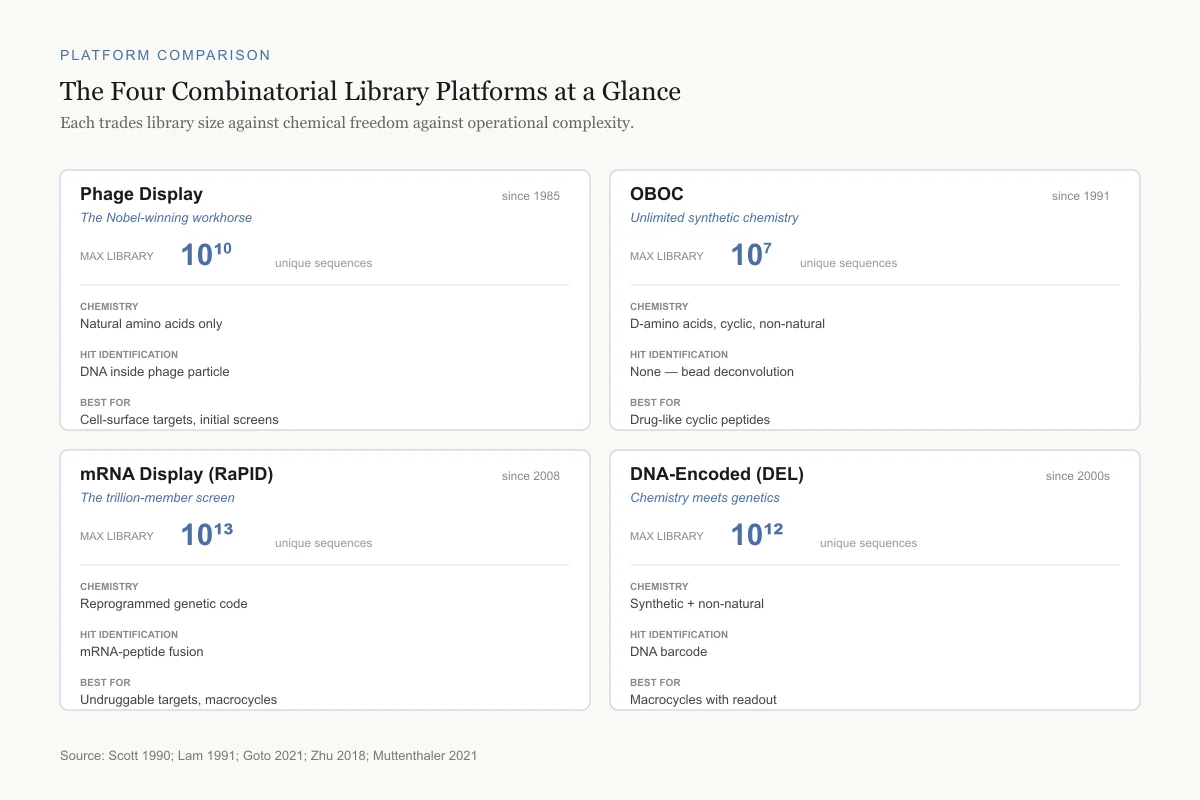
Platform Comparison
The Four Combinatorial Library Platforms at a Glance
Each trades library size against chemical freedom against operational complexity.
Phage Display
since 1985The Nobel-winning workhorse
Chemistry
Natural amino acids only
Hit identification
DNA inside phage particle
Best for
Cell-surface targets, initial screens
OBOC
since 1991Unlimited synthetic chemistry
Chemistry
D-amino acids, cyclic, non-natural
Hit identification
None — bead deconvolution
Best for
Drug-like cyclic peptides
mRNA Display (RaPID)
since 2008The trillion-member screen
Chemistry
Reprogrammed genetic code
Hit identification
mRNA-peptide fusion
Best for
Undruggable targets, macrocycles
DNA-Encoded (DEL)
since 2000sChemistry meets genetics
Chemistry
Synthetic + non-natural
Hit identification
DNA barcode
Best for
Macrocycles with readout
Source: Scott 1990; Lam 1991; Goto 2021; Zhu 2018; Muttenthaler 2021
 View as image
View as imagePhage display: the technology that won a Nobel Prize
Phage display is the oldest and most validated combinatorial peptide library technology. George Smith developed the core technique in 1985, demonstrating that foreign peptide sequences could be fused to a coat protein of filamentous bacteriophage (M13) and displayed on the virion surface while the encoding DNA remained packaged inside the same particle. This created a direct physical link between genotype and phenotype: each phage particle displays one peptide and carries the gene that encodes it.[4]
Scott and Smith (1990) published the first demonstration of a phage-displayed peptide library in Science. They created a library of millions of random hexapeptide sequences fused to the pIII coat protein of filamentous phage, then selected peptides that bound to a target antibody through iterative rounds of affinity purification (biopanning). The selected phage were amplified in E. coli, and the winning sequences were identified by DNA sequencing.[1]
In the same year, Cwirla et al. independently reported a similar approach in PNAS. Their library contained approximately 3 x 10^8 random hexapeptides displayed on phage pIII, and they demonstrated selection of peptides binding a monoclonal antibody specific for beta-endorphin's N-terminus. The winning peptides shared consensus motifs that matched the natural epitope, validating that the library could rediscover biologically relevant binding sequences from random diversity.[2]
The technology matured rapidly. By the mid-1990s, phage display libraries had been used to discover peptide mimetics of erythropoietin that activated the EPO receptor despite sharing no sequence homology with the natural hormone, demonstrating that combinatorial selection could find entirely novel solutions to molecular binding problems. Smith and Gregory Winter shared the 2018 Nobel Prize in Chemistry for phage display of peptides and antibodies, with the Nobel committee citing the technology's transformative impact on drug discovery and protein engineering.[4]
Modern phage display libraries typically contain 10^9 to 10^10 unique clones. They are relatively inexpensive to construct, selection is straightforward, and the genetic encoding allows easy hit identification. Castel et al. (2011) reviewed phage display's application to antiviral research, documenting how the technology identified peptides targeting influenza hemagglutinin, HIV gp120, hepatitis C virus NS3, and dengue virus envelope protein.[6]
The main limitation of standard phage display is chemical constraint. Because the peptides are produced by the bacterial ribosome, they are limited to the 20 natural L-amino acids in linear or disulfide-constrained formats. No D-amino acids, no backbone modifications, no unnatural side chains, no non-peptide elements. This restricts the chemical space that phage libraries can explore relative to synthetic approaches.
Hampton et al. (2024) reviewed emerging strategies to overcome this limitation in Chemical Reviews. By incorporating noncanonical amino acids through amber codon suppression, chemical post-translational modification of displayed peptides, or hybrid biological-chemical diversification, researchers are expanding phage display's chemical repertoire beyond the natural amino acid alphabet while retaining the genetic encoding advantage.[8]
OBOC libraries: one bead, one compound, unlimited chemistry
Lam et al. introduced the one-bead-one-compound (OBOC) concept in Nature in 1991. The method uses split-and-mix combinatorial synthesis on resin beads. At each coupling step, the bead pool is divided into portions equal to the number of building blocks, each portion is reacted with a different amino acid, and all portions are recombined before the next cycle. The result: each 80-100 micrometer bead carries approximately 100 picomoles of a single peptide sequence, and the library collectively displays millions of distinct compounds.[3]
The key advantage of OBOC over phage display is chemical freedom. Because synthesis is entirely chemical rather than biological, OBOC libraries can incorporate D-amino acids, N-methylated residues, beta-amino acids, cyclization chemistries, and non-peptide building blocks. This opens chemical space that biological display methods cannot access. The resulting compounds are often more drug-like, with improved protease resistance and membrane permeability compared to all-L-amino acid peptides.
Screening is typically done by incubating the bead library with a fluorescently labeled or enzyme-tagged target protein, then identifying positive beads by color change, fluorescence, or other visual readouts under a microscope. Hit beads are physically picked and their sequences determined by mass spectrometry or Edman degradation. Liu et al. (2017) reviewed OBOC's application to tumor-targeting peptide discovery, documenting multiple peptides identified from OBOC screens that showed selective binding to cancer cells in vitro and tumor accumulation in vivo.[7]
OBOC's limitations are scale and throughput. Practical library sizes max out at approximately 10^6 to 10^8 compounds, several orders of magnitude smaller than phage or mRNA display. Screening is lower-throughput (visual bead picking versus bulk selection), and hit identification by mass spectrometry requires specialized equipment. There is also no genetic encoding, so the link between structure and identity depends on deconvolution methods that can be technically challenging.
Despite these constraints, OBOC has produced clinically relevant hits in areas where chemical diversity matters more than library size, particularly for cyclic peptides, peptidomimetics, and compounds targeting intracellular protein-protein interactions where cell penetration requires non-natural structural features.
mRNA display and the RaPID system: trillion-member libraries
mRNA display achieves the largest library diversities of any peptide screening technology. The technique, developed in the late 1990s, creates a covalent bond between each peptide and the mRNA that encodes it by attaching puromycin to the mRNA's 3' end. During translation, the ribosome incorporates the puromycin into the growing peptide chain, physically linking genotype and phenotype. The resulting mRNA-peptide fusions can be selected against a target, and the mRNA from winning molecules is reverse-transcribed, amplified by PCR, and fed back into the next selection round.[5]
Goto and Suga (2021) described the RaPID (Random nonstandard Peptides Integrated Discovery) system in Accounts of Chemical Research. RaPID combines mRNA display with genetic code reprogramming, a technique that reassigns sense codons to nonproteinogenic amino acids during in vitro translation. This allows RaPID libraries to contain macrocyclic peptides incorporating D-amino acids, N-methyl amino acids, and other non-natural building blocks, all while maintaining the mRNA-based genotype-phenotype link that enables selection from trillion-member libraries.[5]
The numbers are striking. A single RaPID experiment typically screens 10^12 to 10^13 unique macrocyclic peptide sequences. By comparison, a large pharmaceutical company's small molecule screening collection contains approximately 10^6 compounds. RaPID thus accesses a chemical diversity six to seven orders of magnitude larger than conventional high-throughput screening.
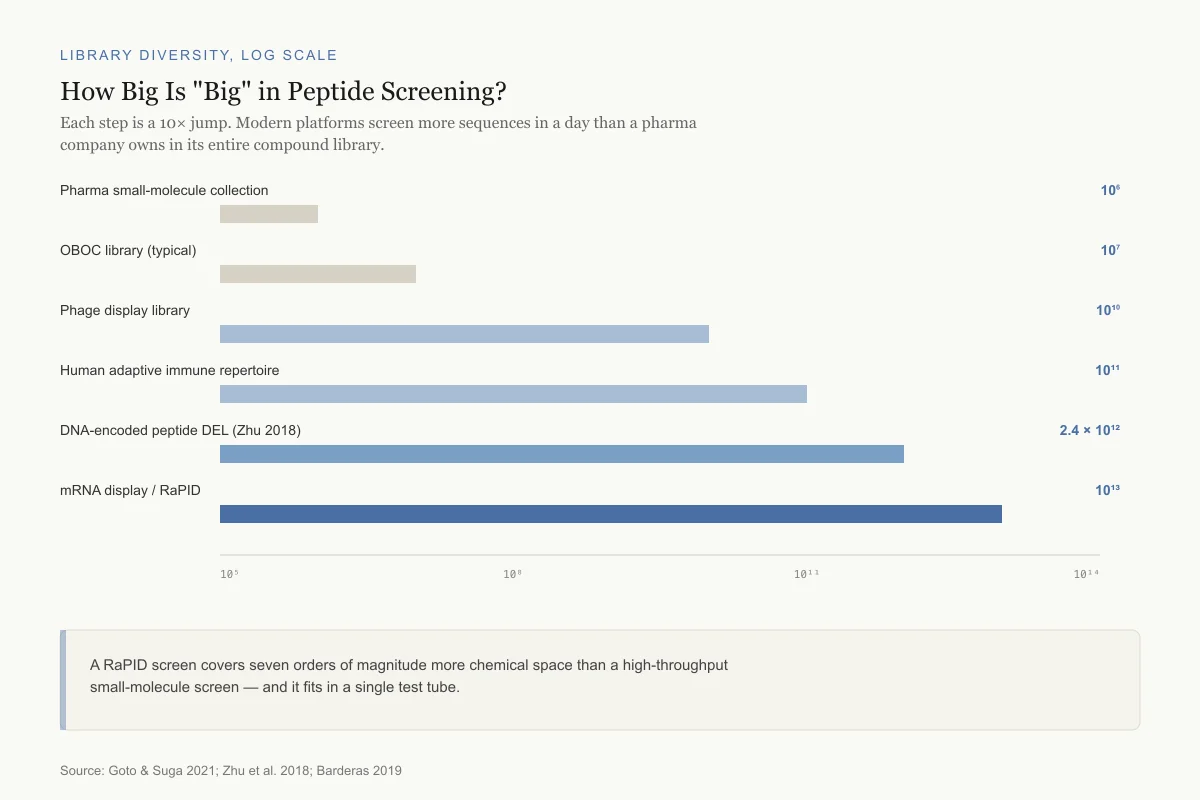
Library Diversity, Log Scale
How Big Is "Big" in Peptide Screening?
Each step on the axis is a 10× jump. Modern platforms screen more sequences in a day than a pharma company owns in its entire compound library.
Pharma small-molecule collection
10⁶OBOC library (typical)
10⁷Phage display library
10¹⁰Human adaptive immune repertoire
10¹¹DNA-encoded peptide DEL (Zhu 2018)
2.4 × 10¹²mRNA display / RaPID
10¹³A RaPID screen covers seven orders of magnitude more chemical space than a high-throughput small-molecule screen — and it fits in a single test tube.
Source: Goto & Suga 2021; Zhu et al. 2018; Barderas 2019
 View as image
View as imageThe system has identified potent binders against a growing list of targets previously considered undruggable by small molecules, including protein-protein interactions, allosteric sites, and conformational epitopes. Many of these macrocyclic peptide hits achieve dissociation constants in the low nanomolar to subnanomolar range, rivaling monoclonal antibodies in affinity while remaining small enough (typically 1-2 kDa) to potentially access intracellular targets.
The limitations of mRNA display are cost and complexity. The in vitro translation, genetic code reprogramming, and selection infrastructure require specialized expertise and reagents. Library construction is more technically demanding than phage display. And while the selected peptides demonstrate strong binding in biochemical assays, translating that binding into cell-active and in vivo-active compounds remains the primary bottleneck. Macrocyclic peptides generally have limited oral bioavailability and cell permeability, though N-methylation and other structural features accessible through RaPID can improve these properties.
DNA-encoded peptide libraries: chemistry meets genetics
DNA-encoded libraries (DELs) represent a hybrid approach that attaches DNA barcodes to chemically synthesized compounds. For peptide DELs, each synthesis step adds both a building block and a corresponding DNA tag, creating a molecular record of each compound's structure. After affinity selection against an immobilized target, the DNA tags from bound compounds are amplified and sequenced, revealing the chemical identities of the hits.
Zhu et al. (2018) published the design and application of a DNA-encoded macrocyclic peptide library in ACS Chemical Biology. Their library contained 2.4 x 10^12 members composed of 4 to 20 natural and non-natural amino acids, cyclized through various chemistries. They performed affinity-based selection against two therapeutic targets, VHL (a cancer-relevant E3 ubiquitin ligase) and RSV N protein (a respiratory syncytial virus target), identifying selective binders from the enormous chemical space.[9]
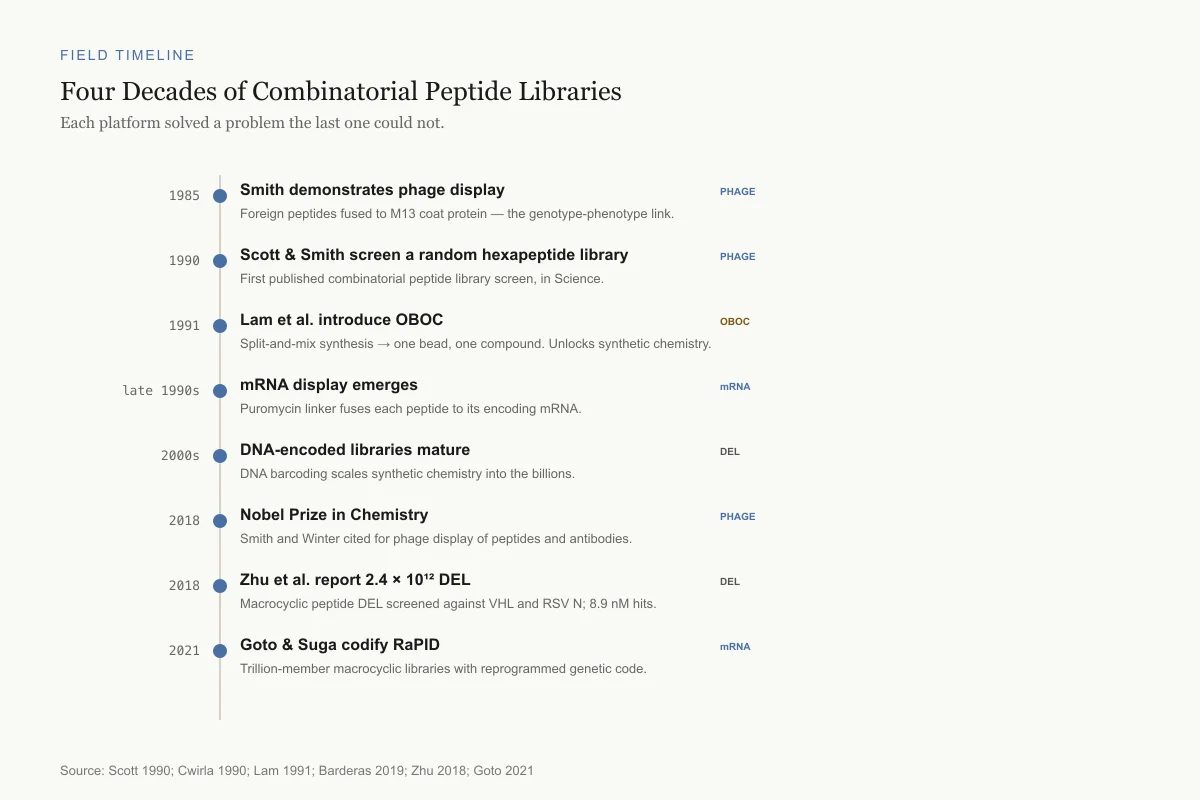
Field Timeline
Four Decades of Combinatorial Peptide Libraries
Each platform solved a problem the last one could not.
Smith demonstrates phage display
PhageForeign peptides fused to M13 coat protein — the genotype-phenotype link.
Scott & Smith screen a random hexapeptide library
PhageFirst published combinatorial peptide library screen, in Science.
Lam et al. introduce OBOC
OBOCSplit-and-mix synthesis → one bead, one compound. Unlocks synthetic chemistry.
mRNA display emerges
mRNAPuromycin linker fuses each peptide to its encoding mRNA.
DNA-encoded libraries mature
DELDNA barcoding scales synthetic chemistry into the billions.
Nobel Prize in Chemistry
PhageSmith and Winter cited for phage display of peptides and antibodies.
Zhu et al. report 2.4 × 10¹² DEL
DELMacrocyclic peptide DEL screened against VHL and RSV N; 8.9 nM hits.
Goto & Suga codify RaPID
mRNATrillion-member macrocyclic libraries with reprogrammed genetic code.
Source: Scott 1990; Cwirla 1990; Lam 1991; Barderas 2019; Zhu 2018; Goto 2021
 View as image
View as imageDELs offer a unique combination of large library size (approaching mRNA display scale) with the chemical flexibility of synthetic chemistry (approaching OBOC diversity). They can incorporate non-natural amino acids, diverse cyclization chemistries, and non-peptide elements while maintaining genetic encoding for hit identification. The DNA barcode also enables quantitative analysis of enrichment patterns across the entire library, revealing structure-activity relationships that single-compound testing would miss.
The main limitation is that DEL selection occurs on the DNA-peptide conjugate, meaning the DNA tag is present during binding. This can introduce artifacts if the DNA interacts with the target. Additionally, the synthesis yield and purity of individual library members in a DEL are lower than in dedicated OBOC synthesis, which can complicate hit validation.
Platform comparison: choosing the right tool
| Feature | Phage Display | OBOC | mRNA Display (RaPID) | DEL |
|---|---|---|---|---|
| Library size | 10^9-10^10 | 10^6-10^8 | 10^12-10^13 | 10^9-10^12 |
| Chemical diversity | Natural amino acids only (standard) | Unlimited (synthetic) | Expanded via code reprogramming | Broad (synthetic + encoded) |
| Genotype-phenotype link | DNA inside phage | None (deconvolution) | mRNA-peptide fusion | DNA barcode |
| Amplification | Biological (phage growth) | None | PCR | PCR |
| Cyclic peptides | Disulfide only (standard) | Any chemistry | Thioether, other | Any chemistry |
| Equipment needs | Standard molecular biology | Microscopy, mass spec | Specialized translation system | Standard molecular biology + synthesis |
| Track record | 35+ years, Nobel Prize | 35+ years | 15+ years | 10+ years |
No single platform dominates. Phage display remains the most accessible and best-validated approach for initial target engagement studies. OBOC excels when chemical diversity matters more than library size. mRNA display (particularly RaPID) is the choice when maximizing library size and accessing macrocyclic chemical space are priorities. DELs offer a middle ground with both genetic encoding and synthetic flexibility.
In practice, drug discovery campaigns increasingly use multiple platforms sequentially or in parallel. A phage display screen might identify a linear peptide hit, which is then optimized using OBOC-derived cyclic analogs, with mRNA display providing the largest possible chemical space search when initial approaches fail to find potent enough leads.
From library hit to drug: the translational gap
Finding a peptide that binds a target in a test tube is the beginning, not the end, of drug discovery. Library hits must be validated for specificity, optimized for potency, and then engineered for drug-like properties including stability, cell permeability, and pharmacokinetics.
Muttenthaler et al. (2021) reviewed trends in peptide drug discovery in Nature Reviews Drug Discovery, noting that combinatorial library screening has become one of three major pipelines for peptide lead discovery alongside rational design from natural hormones and isolation from venoms and other natural sources. Over 80 peptide drugs have reached the market across diverse therapeutic areas including diabetes (GLP-1 agonists), cancer (luteinizing hormone-releasing hormone analogs), osteoporosis (parathyroid hormone analogs), and infectious disease.[10]
The translational bottleneck for library-derived peptides is often oral bioavailability. Most peptide library hits are administered by injection because they are degraded in the gastrointestinal tract and do not cross cell membranes efficiently. Macrocyclic peptides from RaPID and OBOC libraries, which can incorporate N-methylation and other protease-resistant features, are pushing this boundary. Several macrocyclic peptide drug candidates with oral bioavailability have entered clinical trials, though none from library screening has yet reached approval via the oral route.
The growing intersection of combinatorial libraries with artificial intelligence is accelerating the optimization step. Machine learning models trained on library screening data can predict binding affinity, selectivity, and drug-like properties for unscreened sequences, effectively expanding the useful chemical space beyond what is physically screened. This computational amplification of library data is becoming a standard component of modern peptide drug discovery campaigns.
Applications across therapeutic areas
Combinatorial peptide libraries have found applications far beyond the antibody-binding studies that launched the field. Liu et al. (2017) documented the use of phage display and OBOC libraries to identify tumor-targeting peptides, including RGD-containing sequences that bind integrins on tumor vasculature and peptides that home specifically to particular tumor types in vivo. Several of these have advanced to clinical imaging agents and drug delivery vehicles for anticancer peptides.[7]
Castel et al. (2011) reviewed phage display applications in antiviral research, where library-derived peptides have been identified against HIV, influenza, hepatitis B and C, and dengue targets. Many of these peptides block viral entry by mimicking receptor-binding domains or by targeting conserved fusion machinery.[6]
In the antimicrobial peptide space, library screening has identified synthetic sequences with potent activity against drug-resistant bacteria, including candidates that disrupt bacterial membranes through mechanisms distinct from conventional antibiotics. The ability of OBOC and RaPID libraries to incorporate D-amino acids and cyclic structures is particularly valuable here, as these modifications confer resistance to bacterial proteases that would rapidly degrade natural L-amino acid peptides.
Beyond therapeutics, combinatorial peptide libraries are widely used for diagnostic development (identifying peptides that bind disease biomarkers), material science (discovering self-assembling peptide sequences), and basic research (mapping protein-protein interaction interfaces).
What remains unsolved
Despite four decades of development and a Nobel Prize, combinatorial peptide libraries face several persistent challenges.
The oral bioavailability problem. Most library-derived peptides cannot be taken as pills. Cyclic and N-methylated peptides from RaPID and OBOC libraries show improved oral absorption in some cases, but no general solution exists. The physicochemical properties that make a peptide a good target binder (large surface area, many hydrogen bond donors) are the same properties that limit membrane permeability.
The cell penetration barrier. Many high-value drug targets, including transcription factors and intracellular protein-protein interactions, are inside cells. Library-derived peptides typically bind extracellular or surface targets. Conjugation with cell-penetrating peptides is one approach, but systematic solutions for intracellular delivery of macrocyclic peptides are still in development.
Library bias. No library is truly random. Phage display is biased by codon usage, toxicity of certain displayed sequences, and E. coli growth rates. OBOC libraries are biased by coupling efficiencies of different building blocks. mRNA display is biased by translation efficiency. Understanding and correcting these biases is essential for ensuring that the best possible binders are represented in the library and not lost to technical artifacts.
Integration with computational methods. AI-driven peptide design is advancing rapidly, but most computational models are trained on relatively small datasets compared to the diversity that combinatorial libraries can generate. Better integration between experimental library screening and computational prediction could dramatically reduce the number of screening rounds needed to find optimized drug candidates.
The Bottom Line
Combinatorial peptide libraries have transformed drug discovery from a rational design exercise into an evolutionary search across chemical space. Four platforms, phage display (10^10 diversity), OBOC (unlimited chemistry), mRNA display (10^12+ scale), and DNA-encoded libraries (genetic encoding plus synthetic flexibility), each address different aspects of the diversity-chemistry-throughput tradeoff. The field has produced a Nobel Prize, contributed to over 80 approved peptide drugs, and continues to expand through integration of noncanonical amino acids, macrocyclic architectures, and AI-driven optimization. The remaining frontiers are oral bioavailability, intracellular delivery, and deeper computational integration.
Sources & References
- 1RPEP-00167·Scott, J K et al. (1990). “Searching for peptide ligands with an epitope library..” Science (New York.Study breakdown →PubMed →↩
- 2RPEP-00151·Cwirla, S E et al. (1990). “The 1990 Paper That Launched Phage Display Peptide Libraries.” Proceedings of the National Academy of Sciences of the United States of America.Study breakdown →PubMed →↩
- 3RPEP-00199·Lam, K S et al. (1991). “The 1991 Nature Paper That Invented One-Bead-One-Peptide Libraries for Drug Discovery.” Nature.Study breakdown →PubMed →↩
- 4RPEP-04070·Barderas, Rodrigo et al. (2019). “The 2018 Nobel Prize in Chemistry: phage display of peptides and antibodies..” Analytical and bioanalytical chemistry.Study breakdown →PubMed →↩
- 5RPEP-05415·Goto, Yuki et al. (2021). “The RaPID Platform for the Discovery of Pseudo-Natural Macrocyclic Peptides..” Accounts of chemical research.Study breakdown →PubMed →↩
- 6RPEP-01744·Castel, Guillaume et al. (2011). “Phage display of combinatorial peptide libraries: application to antiviral research..” Molecules (Basel.Study breakdown →PubMed →↩
- 7RPEP-03373·Liu, Ruiwu et al. (2017). “Tumor-targeting peptides from combinatorial libraries..” Advanced drug delivery reviews.Study breakdown →PubMed →↩
- 8RPEP-08339·Hampton, J Trae et al. (2024). “Diversification of Phage-Displayed Peptide Libraries with Noncanonical Amino Acid Mutagenesis and Chemical Modification..” Chemical reviews.Study breakdown →PubMed →↩
- 9RPEP-04020·Zhu, Zhengrong et al. (2018). “Design and Application of a DNA-Encoded Macrocyclic Peptide Library..” ACS chemical biology.Study breakdown →PubMed →↩
- 10RPEP-05633·Muttenthaler, Markus et al. (2021). “A Century of Peptide Drugs: How 80+ Medicines Went from Insulin to Semaglutide.” Nature reviews. Drug discovery.Study breakdown →PubMed →↩