De Novo Peptide Design: Building Drugs with AI
Computational Peptide Design
94% Hit Rate
An AMP-specific large language model called AMP Designer generated 18 de novo antimicrobial peptides in 48 days. When synthesized and tested, 17 of 18 were active against target bacteria.
AMP Designer, 2025
AMP Designer, 2025
If you only read one thing
De novo peptide design means using computers to invent brand-new peptide medicines instead of searching for them. The possible sequences are astronomical — a 20-amino-acid peptide has more possible versions than there are atoms in the known universe. Modern AI tools like AlphaFold and peptide language models can pick a handful of promising designs out of that ocean; in one recent test, 17 of 18 AI-designed antibiotic peptides actually worked in the lab. The technology is real and fast, but no AI-designed peptide has yet become an approved drug. Think of it as the most exciting early chapter of a long book.
A 10-amino-acid peptide has 20 to the power of 10 possible sequences: over 10 trillion combinations. A 20-amino-acid peptide has more possible sequences than atoms in the observable universe. Traditional drug discovery, which relies on screening natural peptide libraries or systematically modifying known sequences, can explore only a vanishingly small fraction of this space. De novo peptide design uses computational methods to generate entirely new peptide sequences that have never existed in nature, targeting specific biological activities. Artificial intelligence has transformed this field. A 2025 review in Drug Discovery Today described how deep generative models are now enabling the design of target-specific peptide binders that would take years to discover through conventional methods.[1] No AI-designed peptide drug has reached market approval yet, but the pipeline from computation to laboratory validation is accelerating at a pace that has changed the economics of peptide drug development.
Key Takeaways
- An AMP-specific language model (AMP Designer) generated 18 de novo antimicrobial peptides in 48 days; 17 of 18 (94.4%) showed activity when synthesized and tested
- A Feedback Generative Adversarial Network (FBGAN) with protein language model classifiers achieved comparable or superior performance to established AMP design methods (Zervou et al., IJMS, 2024)
- AlphaFold 3 can now predict protein-peptide interactions with 50% higher accuracy than its predecessor, enabling structure-guided peptide design
- The design space for a 20-amino-acid peptide exceeds 10 to the power of 26 possible sequences, making computational approaches essential for efficient exploration
- Deep generative models (GANs, diffusion models, transformers) are the three dominant AI architectures currently used for de novo peptide generation
- No AI-designed peptide therapeutic has received market approval as of 2026, though several are in early-phase clinical trials
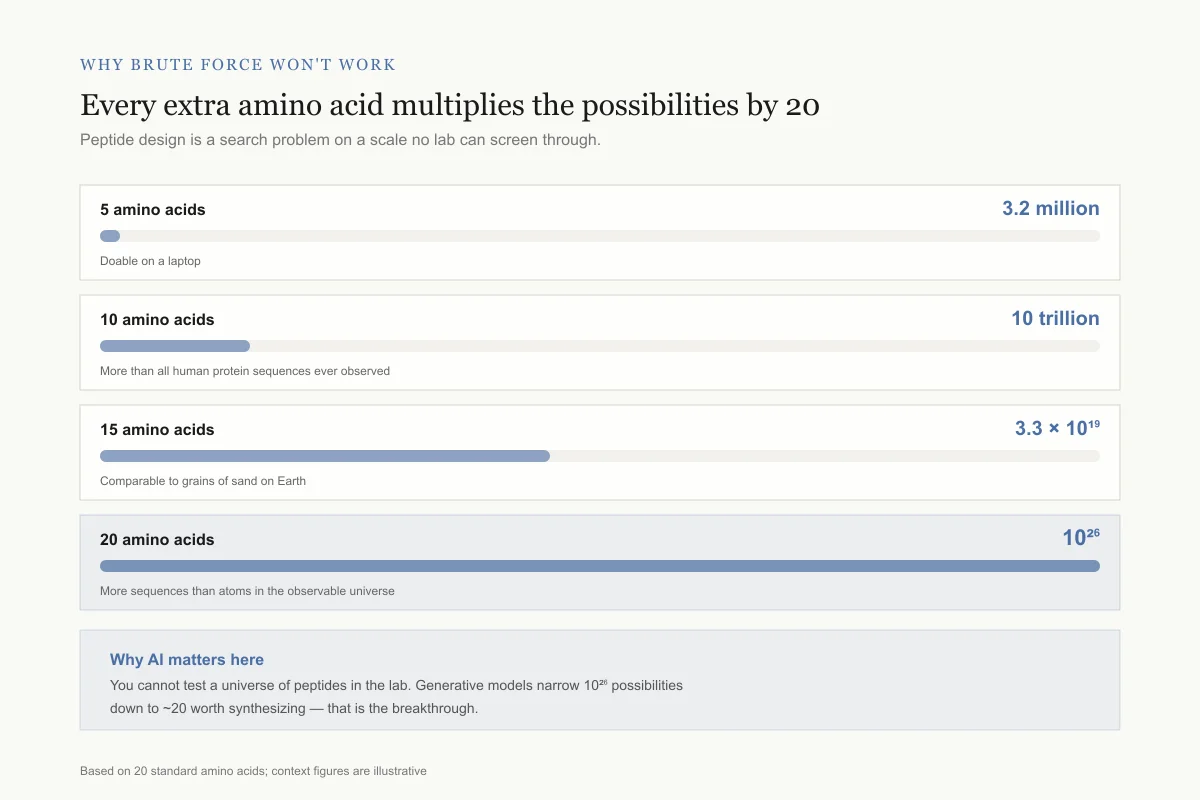
Why brute force won't work
Every extra amino acid multiplies the possibilities by 20
Peptide design is a search problem on a scale no lab can screen through.
5 amino acids
3.2 millionDoable on a laptop
10 amino acids
10 trillionBigger than all human protein sequences ever observed
15 amino acids
3.3 × 10¹⁹Comparable to grains of sand on Earth
20 amino acids
10²⁶More sequences than atoms in the observable universe
Why AI matters here: you cannot test a universe of peptides in the lab. Generative models narrow 10²⁶ possibilities down to ~20 worth synthesizing — that is the breakthrough.
Scale based on 20 standard amino acids; context figures are illustrative
 View as image
View as imageWhy Peptides Are Hard to Design
Peptide drug design faces challenges that differ from both small-molecule and large-protein drug design.
Flexibility: Unlike folded proteins with stable 3D structures, short peptides (under ~30 amino acids) are highly flexible in solution. They sample many conformations, making it difficult to predict which shape they will adopt when binding a target. This flexibility is also what makes peptides useful; they can access protein surfaces that rigid small molecules cannot.
Proteolytic degradation: Natural peptides are rapidly broken down by proteases in the body. A designed peptide must not only bind its target but also survive long enough to reach it. Design strategies include incorporating non-natural amino acids (D-amino acids, N-methylated residues), cyclization (connecting the peptide's ends to form a ring), and stapling (adding chemical bridges to stabilize helical structure).
Membrane permeability: Most peptides cannot cross cell membranes, limiting them to extracellular targets. Cell-penetrating peptides exist but adding cell-penetration properties while maintaining target specificity is a dual optimization problem.
Immunogenicity: Synthetic peptides can trigger immune responses, particularly with repeated administration. Predicting immunogenicity computationally is still unreliable. For more on how this affects compounded peptide products, see Compounded Peptide Safety Monitoring: The Regulatory Gap.
These constraints mean that de novo peptide design is not just about finding a sequence that binds a target. It is a multi-objective optimization problem: binding affinity, selectivity, stability, membrane permeability, solubility, low immunogenicity, and synthetic accessibility must all be considered simultaneously.
The Three Generations of Computational Design
First Generation: Physics-Based Methods
The earliest computational approaches to peptide design relied on molecular mechanics and molecular dynamics simulations. These methods model the physical forces between atoms (electrostatic interactions, van der Waals forces, hydrogen bonds) to predict peptide structures and binding energies. For how these simulations work in detail, see Molecular Dynamics Simulations for Peptides: Watching Molecules Move.
Physics-based methods are accurate but computationally expensive. Simulating a single peptide-protein interaction for microseconds can require days of computing time on specialized hardware. Designing a peptide from scratch, where millions of candidates must be evaluated, was impractical with physics-based methods alone.
Rosetta, developed by David Baker's lab at the University of Washington, represented a significant advance by combining physics-based energy functions with knowledge-based statistical potentials derived from known protein structures. Rosetta could design new protein and peptide structures de novo, and the Baker lab used it to create several novel proteins that were validated experimentally. RosettaFold and later RFdiffusion extended this approach using deep learning.
Second Generation: Machine Learning Classifiers
The second generation moved from simulating physics to learning patterns from data. Machine learning classifiers trained on databases of known bioactive peptides (antimicrobial peptides, cell-penetrating peptides, anticancer peptides) learned to distinguish active from inactive sequences. Given a candidate peptide sequence, these classifiers could predict whether it would likely be active.
The limitation: classifiers can evaluate proposed sequences, but they cannot generate new ones. Researchers still had to propose candidates through mutation, recombination, or random sampling, then use classifiers to filter. This was more efficient than physics-based screening but still explored the sequence space inefficiently.
Sequence-based features (amino acid composition, physicochemical properties, k-mer frequencies) combined with machine learning algorithms (support vector machines, random forests, gradient boosting) could achieve classification accuracies above 90% for well-defined peptide classes. These classifiers remain useful as evaluation filters within modern generative pipelines. For how one amino acid change can transform a peptide's properties, see Structure-Activity Relationships: How Changing One Amino Acid Changes Everything.
Third Generation: Deep Generative Models
The current generation uses deep learning models that both generate new sequences and evaluate their properties. Three architectures dominate:
Generative Adversarial Networks (GANs): Two neural networks compete. A generator creates new peptide sequences; a discriminator evaluates whether they resemble real bioactive peptides. Through iterative training, the generator learns to produce increasingly realistic sequences. Zervou et al. (2024) demonstrated that integrating advanced classifiers (including the ESM2 protein language model) into a Feedback GAN framework for antimicrobial peptide design achieved performance "comparable or even superior" to established methods like AMPGAN and HydrAMP.[2]
Variational Autoencoders (VAEs): These models learn a compressed representation (latent space) of peptide sequences. New peptides are generated by sampling from this latent space and decoding back to sequences. The advantage is that the latent space organizes peptide properties continuously, allowing controlled navigation between different activity profiles.
Transformer Models: The same architecture behind large language models like GPT. Trained on peptide sequence databases, transformers learn the "grammar" of bioactive peptides and can generate new sequences that follow these learned patterns. Protein language models like ESM2 (trained on millions of protein sequences) provide powerful representations that can be adapted for peptide design. The AMP Designer, a peptide-specific language model, generated 18 de novo antimicrobial peptides in 48 days with a 94.4% hit rate when synthesized and tested.
Diffusion Models: The newest architecture, adapted from image generation (like DALL-E and Stable Diffusion). RFdiffusion, from the Baker lab, generates entirely new protein and peptide structures by iteratively denoising random structures. AMPGen combined latent diffusion with protein language models to produce structurally diverse antimicrobial peptides with over 80% antibacterial activity when synthesized.
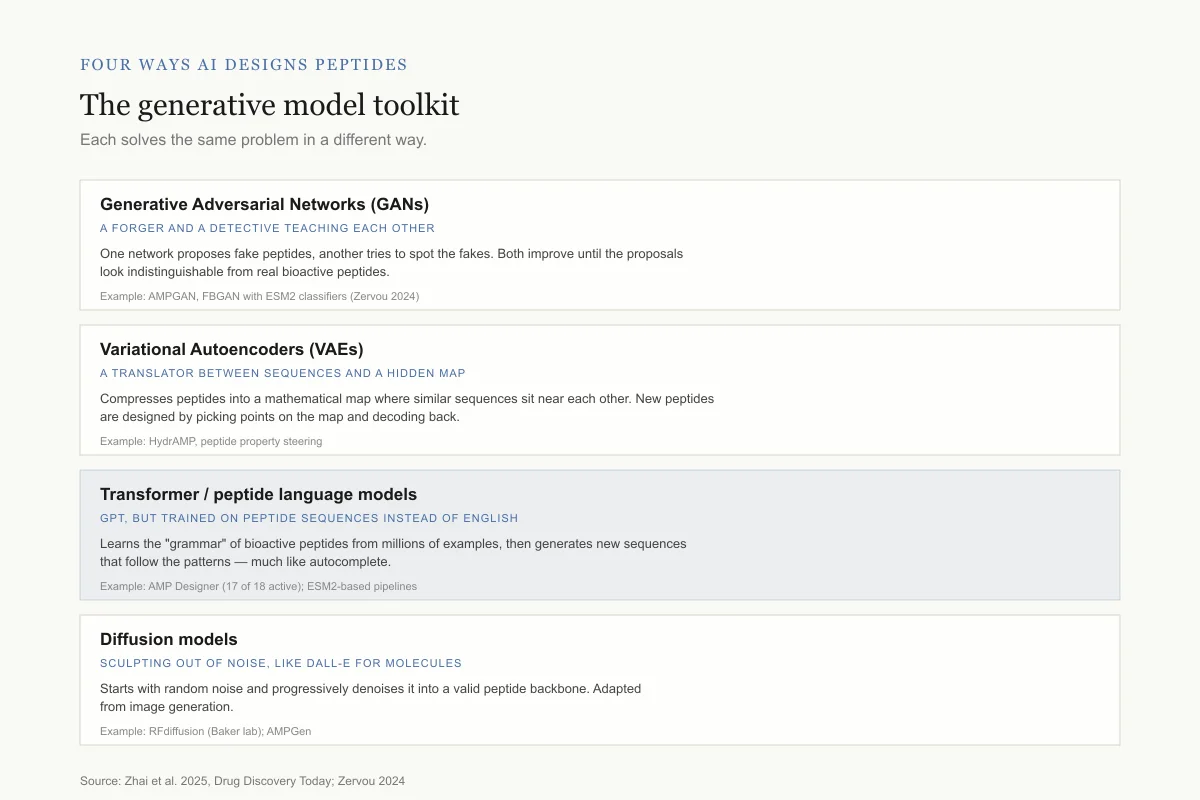
Four ways AI designs peptides
The generative model toolkit
Each approach solves the same problem — generate novel sequences predicted to work — in a different way.
Generative Adversarial Networks (GANs)
A forger and a detective teaching each other
One network proposes fake peptides, another tries to spot the fakes. Both improve until the proposals look indistinguishable from real bioactive peptides.
Best for: Generates realistic, novel sequences quickly
Example: AMPGAN, FBGAN with ESM2 classifiers (Zervou 2024)
Variational Autoencoders (VAEs)
A translator between sequences and a hidden map
Compresses peptides into a mathematical map where similar sequences sit near each other. New peptides are designed by picking points on the map and decoding back.
Best for: Smooth control over peptide properties
Example: HydrAMP, peptide property steering
Transformer / peptide language models
GPT, but trained on peptide sequences instead of English
Learns the "grammar" of bioactive peptides from millions of examples, then generates new sequences that follow the patterns — much like autocomplete.
Best for: Extremely high hit rates when trained on rich data
Example: AMP Designer (17 of 18 active); ESM2-based pipelines
Diffusion models
Sculpting out of noise, like DALL-E for molecules
Starts with random noise and progressively denoises it into a valid peptide backbone. Adapted from image generation.
Best for: Produces diverse, novel 3D structures
Example: RFdiffusion (Baker lab); AMPGen
In practice: modern pipelines combine these. A transformer might propose candidates, a diffusion model refines 3D structure, and AlphaFold 3 predicts how they'll bind the target — all before anything is synthesized.
Source: Zhai et al. 2025, Drug Discovery Today; Zervou 2024
 View as image
View as imageAlphaFold and Structure-Guided Design
AlphaFold, developed by DeepMind, won the 2024 Nobel Prize in Chemistry for solving protein structure prediction. AlphaFold 3 expanded beyond single protein structures to predict protein-protein and protein-peptide interactions with 50% higher accuracy than AlphaFold 2.
For peptide design, AlphaFold's impact is indirect but profound. Before AlphaFold, designing a peptide to bind a specific protein target required knowing that target's 3D structure, usually obtained through X-ray crystallography or cryo-EM (months of experimental work). AlphaFold can predict the target structure computationally, making structure-guided peptide design accessible for any protein with a known sequence.
AlphaFold 2 has been specifically adapted for cyclic peptide design, where predicting the closed-ring structure is essential for evaluating binding geometry and stability. Multiple groups have published workflows using AlphaFold-predicted structures as starting points for peptide optimization.
The combination of AlphaFold (for target structure prediction), generative models (for candidate peptide generation), and molecular dynamics (for binding validation) creates a fully computational pipeline that can go from a protein target to a set of candidate peptide drugs without any experimental structure determination. Whether these computationally designed candidates survive laboratory validation remains the key test.
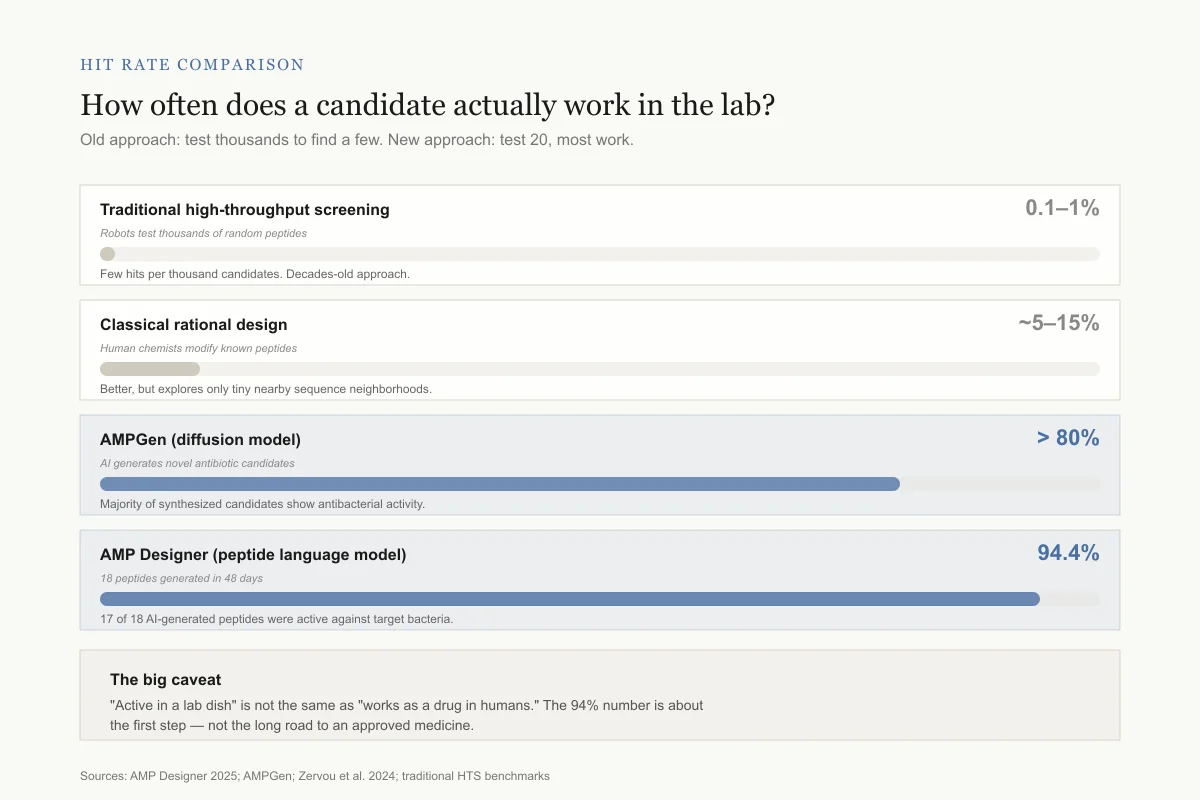
Hit rate comparison
How often does a candidate actually work in the lab?
Old approach: test thousands to find a few. New approach: test twenty and most of them work.
Traditional high-throughput screening
0.1–1%Robots test thousands of random peptides
Few hits per thousand candidates. Decades-old approach.
Classical rational design
~5–15%Human chemists modify known peptides
Better, but explores only tiny nearby sequence neighborhoods.
AMPGen (diffusion model)
> 80%AI generates novel antibiotic candidates
Majority of synthesized candidates show antibacterial activity.
AMP Designer (peptide language model)
94.4%18 peptides generated in 48 days
17 of 18 AI-generated peptides were active against target bacteria.
The big caveat: "active in a lab dish" is not the same as "works as a drug in humans." The 94% number is about the first step — binding bacteria in a test tube — not the long road to an approved medicine.
Sources: AMP Designer 2025; AMPGen; Zervou et al. 2024; traditional HTS benchmarks
 View as image
View as imageThe AI-Lab Loop: From Computation to Synthesis
The most successful de novo peptide design programs operate in an iterative cycle:
- Generate: A generative model proposes hundreds or thousands of candidate peptide sequences
- Filter: Computational classifiers and physics-based scoring functions evaluate candidates for predicted activity, stability, solubility, and synthesizability
- Synthesize: The top candidates (typically 10-50) are synthesized using solid-phase peptide synthesis
- Test: Synthesized peptides are tested in laboratory assays for the target activity
- Learn: Results from testing are fed back into the generative model, improving its predictions for the next cycle
This cycle compresses the traditional drug discovery timeline. Where conventional peptide optimization might take years of iterative synthesis and testing, the AI-lab loop can complete multiple cycles in months. The AMP Designer's 48-day timeline from model output to synthesized, tested peptides represents the current speed frontier.
The 94.4% hit rate (17 of 18 active peptides) is remarkable compared to traditional high-throughput screening, where hit rates of 0.1-1% are typical. Even accounting for the difference between "active in a lab assay" and "effective as a drug," this efficiency gain transforms the economics of peptide drug discovery. For how large virtual libraries are screened computationally, see Virtual Screening of Peptide Libraries: Testing Millions Without a Lab.
Current Applications
Antimicrobial Peptides
Antimicrobial peptide (AMP) design is the most active application area for de novo peptide design. The antibiotic resistance crisis creates urgent demand for new antimicrobial agents, and AMPs offer a mechanism of action (membrane disruption) that is inherently difficult for bacteria to develop resistance against. AI-designed AMPs have shown broad-spectrum activity against ESKAPE pathogens (the six most dangerous antibiotic-resistant bacteria) and multidrug-resistant fungi. For the broader context of AMP research, see Antimicrobial Peptides as Alternatives to Antibiotics: Can They Solve Resistance?.
Protein-Protein Interaction Inhibitors
Many diseases are driven by abnormal protein-protein interactions (PPIs) that small molecules cannot disrupt. Peptides can bind to the large, flat protein surfaces where these interactions occur. AI-based design of PPI inhibitors is particularly promising for targets in cancer (p53-MDM2 interaction), neurodegeneration, and immune signaling.
Targeted Drug Delivery
Cell-penetrating peptides designed computationally can carry therapeutic cargo (drugs, nucleic acids, nanoparticles) across cell membranes to intracellular targets. Tumor-homing peptides can deliver cytotoxic agents specifically to cancer cells, reducing systemic side effects. The design challenge here is dual: the peptide must both penetrate membranes efficiently and maintain specificity for the target cell type. Computational methods can optimize both properties simultaneously by treating them as separate objectives in a multi-objective optimization framework.
Cyclic cell-penetrating peptides designed using AlphaFold-guided workflows have shown improved stability and cellular uptake compared to their linear counterparts. The cyclization constrains the peptide's conformational flexibility, making both structure prediction and design more tractable for computational methods.
Peptide Vaccines
De novo design of peptide epitopes for vaccines enables rapid response to emerging pathogens. Computational methods can predict which viral protein fragments will elicit the strongest immune response, then optimize those fragments for stability and immunogenicity. During the COVID-19 pandemic, computational peptide epitope prediction was used to identify SARS-CoV-2 spike protein regions most likely to generate neutralizing antibody responses, accelerating vaccine antigen selection.
For cancer vaccines, neoantigen peptide design uses patient-specific tumor mutation data to design personalized peptide vaccines that train the immune system to recognize each patient's unique tumor markers. AI models predict which mutant peptide sequences will bind most strongly to the patient's specific MHC (major histocompatibility complex) molecules, a prerequisite for triggering a T-cell immune response. This represents perhaps the most personalized application of de novo peptide design.
Cyclic Peptide Drug Design
Cyclic peptides occupy a unique "middle space" between small molecules and large biologics. They can bind protein surfaces like antibodies but are small enough to potentially cross cell membranes. Their cyclized structure provides metabolic stability superior to linear peptides. Several FDA-approved drugs are cyclic peptides (cyclosporine, daptomycin, octreotide), validating the drug class.
Computational cyclic peptide design is particularly challenging because the closed ring constrains the conformational space differently from linear peptides. Methods combining Rosetta-based backbone sampling with machine learning scoring functions have produced cyclic peptide designs with experimentally validated binding to difficult targets. The field is advancing rapidly with AlphaFold-guided approaches specifically adapted for predicting cyclic peptide structures and their interactions with protein targets.
The Companies and Labs Leading the Field
Several organizations are at the forefront of AI-driven peptide design, each approaching the problem from different angles.
David Baker Lab / Institute for Protein Design (University of Washington): The Baker lab developed Rosetta, RoseTTAFold, and RFdiffusion, the structural biology tools that underpin much of modern computational protein and peptide design. Baker shared the 2024 Nobel Prize in Chemistry for computational protein design. The lab's open-source tools have been widely adopted by academic groups and biotech companies worldwide.
DeepMind (Google): AlphaFold and AlphaFold 3 provide the structure prediction backbone for much of peptide design. Demis Hassabis and John Jumper shared the 2024 Nobel with Baker for their contributions to protein structure prediction.
Generate Biomedicines: Uses generative AI to design protein therapeutics, including peptide-based drugs. Their GB-0669 monoclonal antibody (designed using generative methods) entered Phase 1 clinical trials, demonstrating that AI-designed biologics can reach human testing.
Insilico Medicine: While primarily focused on small molecules, Insilico demonstrated the AI-to-clinic pipeline with INS018_055, the first fully AI-designed drug to enter Phase II trials (for idiopathic pulmonary fibrosis). Their approach to generative chemistry is being extended to peptide-like molecules.
Evotec: Combines AI-driven drug design with high-throughput experimental validation. Their peptide programs use machine learning to optimize both binding affinity and drug-like properties simultaneously.
Academic groups in AMP design: Multiple university groups (including teams in Greece, China, South Korea, and the US) have published open-source tools for antimicrobial peptide design. The competitive landscape in AMP design is one of the most active in computational biology, driven by the antibiotic resistance crisis.
The field is notable for its open-source culture. Unlike many areas of drug discovery, where proprietary methods dominate, key tools in computational peptide design (AlphaFold, RFdiffusion, ESM protein language models) are freely available. This accelerates progress but also means that the barrier to entry for new groups is primarily expertise and computational resources rather than access to tools.
From Design to Drug: The Clinical Development Challenge
The distance between a computationally designed peptide and an approved drug remains substantial. The design phase, now measured in weeks to months, represents only the beginning of a development process that typically spans 10-15 years for conventional drugs.
After computational design and initial laboratory validation, a candidate peptide must pass through:
Lead optimization: Improving the initial hit's potency, selectivity, stability, and pharmacokinetic properties. This is often where computational design methods provide the most value, as AI-guided optimization can explore modification space more efficiently than manual medicinal chemistry.
Preclinical safety: Toxicology, genotoxicity, reproductive toxicity, and immunogenicity studies in animal models. For peptides, immunogenicity assessment is particularly important because repeated injection of synthetic peptides can trigger anti-drug antibody formation.
Formulation and manufacturing: Scaling peptide synthesis from milligram lab quantities to kilogram manufacturing quantities. Peptide manufacturing costs remain higher than small molecule drugs, though solid-phase peptide synthesis and recombinant expression methods are improving.
Clinical trials: Phase I (safety), Phase II (efficacy and dosing), and Phase III (large-scale efficacy). Each phase typically takes 1-3 years. The total clinical development timeline for peptide drugs averages 7-10 years from first-in-human to approval.
The AI contribution shortens the discovery phase (target identification to lead compound) but does not compress clinical development timelines, which are driven by biology and regulatory requirements rather than computational speed. This is why no AI-designed peptide drug has reached market approval despite several years of active development.
What Has Not Been Solved
Despite the advances, several fundamental problems remain:
The validation bottleneck: Computational methods can generate candidates faster than laboratories can test them. The rate-limiting step has shifted from design to experimental validation. Automated peptide synthesis and high-throughput activity screening are improving but have not kept pace with computational generation speed.
In vivo translation: A peptide that is active in a lab assay may fail in an animal model. A peptide that works in mice may fail in humans. The gap between computational design and clinical efficacy remains wide. No AI-designed peptide therapeutic has received market approval as of 2026, though several are in preclinical or early clinical stages.
Multi-objective optimization: Current generative models optimize primarily for predicted bioactivity. Simultaneously optimizing for stability, solubility, membrane permeability, low immunogenicity, and synthetic accessibility is technically possible but dramatically increases model complexity and reduces hit rates.
Training data bias: Generative models learn from databases of known peptides. If the training data is biased toward certain peptide families (abundant antimicrobial peptides, fewer anticancer or anti-inflammatory peptides), the model generates sequences that resemble known peptides rather than truly exploring novel sequence space. Addressing this requires either larger, more diverse training sets or model architectures that can extrapolate beyond their training distribution.
Intellectual property: Traditional drug patents protect specific chemical structures. AI-generated peptides raise questions about inventorship (can an AI be an inventor?), the scope of claims when thousands of candidates can be generated rapidly, and the patentability of computationally obvious sequences. Patent offices worldwide are still developing frameworks for AI-generated inventions, and the outcome will significantly influence investment in computational peptide design.
Reproducibility: Published de novo design methods often report impressive metrics on their own test sets, but reproducing these results on new targets or in different laboratories has proven inconsistent. Standardized benchmarks for evaluating peptide design methods are being developed but are not yet widely adopted.
The Bottom Line
De novo peptide design has progressed from physics-based simulations to deep generative models that can produce bioactive peptides with hit rates above 90%. AlphaFold, diffusion models, transformers, and GANs have created a computational pipeline from protein target to candidate drug that operates in weeks rather than years. Antimicrobial peptides are the most advanced application area, with multiple AI-designed AMPs showing laboratory activity against resistant pathogens. The gap between computational design and clinical translation remains the primary unsolved challenge: no AI-designed peptide drug has reached market approval, and in vivo validation, multi-objective optimization, and training data bias continue to limit the technology.
Sources & References
- 1RPEP-14472·Zhai, Silong et al. (2025). “Artificial intelligence in peptide-based drug design..” Drug discovery today.Study breakdown →PubMed →↩
- 2RPEP-09629·Zervou, Michaela Areti et al. (2024). “Smarter AI Creates Better Antimicrobial Peptide Candidates Using Protein Language Models.” International journal of molecular sciences.Study breakdown →PubMed →↩