Cyclotides: Plant Peptides That Destroy Viral Envelopes
Membrane-Disrupting Antiviral Peptides
2.5 μM anti-HIV EC₅₀
The cyclotide kalata B1 inhibited HIV-1 at 2.5 micromolar concentrations by selectively targeting phosphatidylethanolamine-rich viral membranes, a mechanism distinct from any approved antiretroviral drug.
Henriques et al., Journal of Biological Chemistry, 2011
Henriques et al., Journal of Biological Chemistry, 2011
If you only read one thing
Cyclotides are tough little peptides plants use to defend themselves. What makes them special is how tough: they survive boiling water, stomach acid, and enzymes that destroy most other peptides in minutes. One of them, kalata B1, rips apart HIV particles by punching holes in the lipid shell of the virus itself — a trick no approved HIV drug uses. They are still in early research, not in clinics, but their stability has made them a favorite scaffold for designing the next generation of oral peptide drugs.
Plants cannot run from pathogens or mount an adaptive immune response. Instead, they synthesize chemical arsenals. Among the most remarkable weapons in this arsenal are cyclotides: small proteins of 28 to 37 amino acids with a head-to-tail cyclic backbone reinforced by three disulfide bonds arranged in a cystine knot. This combination of circular backbone and knotted disulfide architecture makes cyclotides extraordinarily resistant to heat, chemical denaturation, and enzymatic degradation. Craik et al. defined the cyclotide family and the cyclic cystine knot (CCK) structural motif in 1999, establishing a new class of plant-derived proteins with no precedent in the protein structure database.[4] Since then, over 400 cyclotide sequences have been identified across multiple plant families, with biological activities spanning antimicrobial, insecticidal, anti-HIV, and anticancer effects. Their antiviral activity, driven by selective disruption of lipid membranes, has made them one of the most studied classes of natural antiviral peptides. This article covers cyclotide structure, their mechanism of membrane destruction, the evidence for antiviral activity, and their potential as drug scaffolds. For deeper coverage of specific subtopics, see the articles on broad-spectrum antiviral peptides, lactoferricin's antiviral mechanisms, and melittin's membrane-disrupting activity.
Key Takeaways
- Kalata B1 inhibited HIV-1 infectivity at 2.5 micromolar concentrations by selectively targeting phosphatidylethanolamine-rich membranes on viral particles, without requiring interaction with viral proteins (Henriques et al., J Biol Chem, 2011).[2]
- The cyclic cystine knot motif makes cyclotides resistant to boiling, extremes of pH, and enzymatic degradation, with kalata B1 retaining full biological activity after 1 hour at 100 degrees Celsius.[1]
- Kalata B1 forms multimeric pores in target membranes, with electrophysiology recordings confirming channel-like activity consistent with oligomeric insertion into lipid bilayers (Huang et al., J Biol Chem, 2009).[5]
- Cyclotides function as natural insecticides, reducing Helicoverpa larval growth by over 50% when incorporated into artificial diets at micromolar concentrations (Craik, Toxins, 2012).[3]
- Over 400 cyclotide sequences identified across the Violaceae, Rubiaceae, Cucurbitaceae, Fabaceae, and Solanaceae plant families represent one of the largest classes of circular proteins in nature.[1]
- Cyclotide scaffolds have been engineered to deliver pharmacologically active peptide sequences orally, with grafted cyclotides retaining bioactivity after oral administration in animal models (Hyun et al., Molecules and Cells, 2025).[9]
What defines a cyclotide?
Cyclotides are plant-derived miniproteins distinguished by two structural features that are individually uncommon and, in combination, unique. The first is a head-to-tail cyclic peptide backbone, meaning the N-terminus and C-terminus are joined by a peptide bond, creating a continuous ring with no free ends. The second is a cystine knot: three disulfide bonds arranged so that two of them and their connecting backbone segments form a ring through which the third disulfide bond threads. This topology is called the cyclic cystine knot (CCK).[4]
Craik et al. (1999) formally defined this structural family and coined the term "cyclotide" after characterizing proteins from plants in the Rubiaceae and Violaceae families. The CCK motif creates a molecular cage that locks the peptide into a compact, rigid structure. Unlike linear peptides, which are rapidly unfolded and degraded in biological environments, cyclotides resist boiling, survive passage through the gastrointestinal tract, and withstand proteolytic enzymes that would destroy most peptides within minutes.[4]
De Veer et al. (2019) published the most comprehensive review of cyclotide structure and function in Chemical Reviews, cataloguing over 400 known cyclotide sequences. These fall into two major subfamilies: Mobius cyclotides, which contain a cis-proline residue that introduces a twist in the backbone, and bracelet cyclotides, which lack this twist. A third group, the trypsin inhibitor cyclotides (exemplified by MCoTI-II from Momordica cochinchinensis), shares the CCK motif but has a different evolutionary origin and primary function as protease inhibitors rather than membrane disruptors.[1]
The distribution of cyclotides across plant families tells a story of convergent evolution. They appear in Violaceae (violets), Rubiaceae (coffee family), Cucurbitaceae (squash family), Fabaceae (legumes), and Solanaceae (nightshades). This taxonomic spread suggests the CCK structural solution to the problem of producing stable bioactive peptides has been independently adopted multiple times in plant evolution.
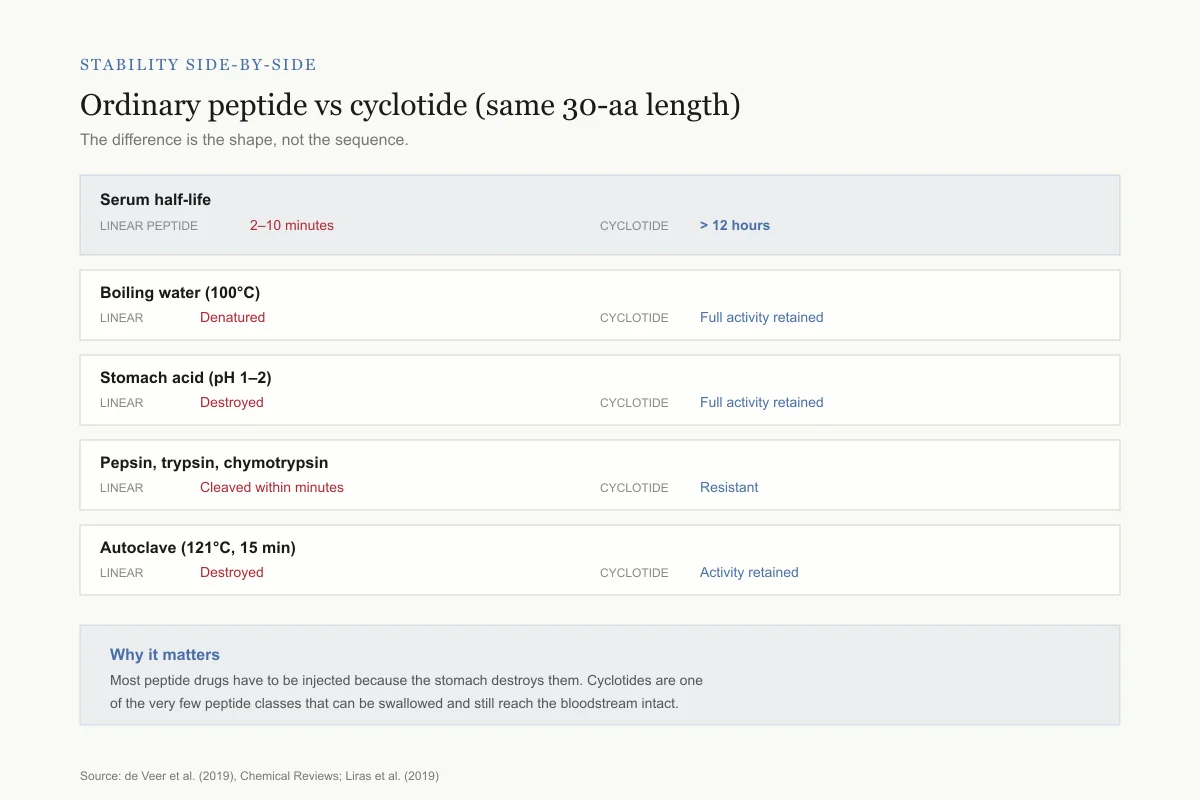
Stability side-by-side
Ordinary 30-aa peptide vs a cyclotide of the same length
Same amino acid count, wildly different survival. The difference is the shape, not the sequence.
Serum half-life
Linear peptide
2–10 minutes
Cyclotide
> 12 hours
Boiling water (100°C)
Linear peptide
Denatured
Cyclotide
Full activity retained
Stomach acid (pH 1–2)
Linear peptide
Destroyed
Cyclotide
Full activity retained
Pepsin, trypsin, chymotrypsin
Linear peptide
Cleaved within minutes
Cyclotide
Resistant
Autoclave (121°C, 15 min)
Linear peptide
Destroyed
Cyclotide
Activity retained
Why it matters: Most peptide drugs have to be injected because the stomach destroys them. Cyclotides are one of the very few peptide classes that can be swallowed and still reach the bloodstream intact.
Source: de Veer et al. (2019), Chemical Reviews; Liras et al. (2019)
 View as image
View as imageHow cyclotides destroy membranes
The antiviral and antimicrobial activities of cyclotides trace to a single mechanism: selective membrane disruption. Cyclotides do not target viral proteins, enzymes, or receptors. They target lipid membranes directly.
Henriques et al. (2011) decoded the membrane selectivity of kalata B1 in a landmark study published in the Journal of Biological Chemistry. They demonstrated that kalata B1's biological activity depends on the lipid composition of target membranes, specifically the presence of phosphatidylethanolamine (PE). When PE-containing lipid vesicles were exposed to kalata B1, the peptide bound rapidly and disrupted membrane integrity. When PE was absent, binding and disruption were minimal.[2]
This PE selectivity has direct relevance to antiviral activity. HIV-1 viral particles are enveloped in a lipid bilayer derived from the host cell plasma membrane during budding. This viral envelope is enriched in PE compared to the outer leaflet of healthy host cell membranes, where PE is predominantly sequestered in the inner leaflet. Kalata B1 can therefore distinguish between healthy host cells and HIV virions based on their lipid composition, destroying the virus while sparing the host. Henriques et al. showed that anti-HIV activity and PE-dependent membrane disruption tracked together across multiple cyclotide variants and lipid compositions.[2]
Huang et al. (2009) revealed the structural mechanism of this disruption. Using dye leakage assays and electrophysiology, they demonstrated that kalata B1 does not simply coat the membrane surface. It inserts into the bilayer and assembles into multimeric pores. Electrophysiological recordings showed channel-like conductance events consistent with the formation of oligomeric peptide pores. The pore model explains how a 30-residue peptide can destroy structures as large as viral particles and bacterial cells: individual peptides self-assemble in the membrane into transmembrane channels that collapse the electrochemical gradient and allow uncontrolled ion and solute flux.[5]
The pore formation process appears to be cooperative. Below a threshold concentration, kalata B1 binds membranes without causing significant leakage. Above the threshold, multiple peptides simultaneously insert and oligomerize, producing catastrophic membrane failure. This concentration-dependent behavior suggests that cyclotides act through a mechanism analogous to the barrel-stave or toroidal pore models described for other antimicrobial peptides, adapted to their unique cyclic topology.
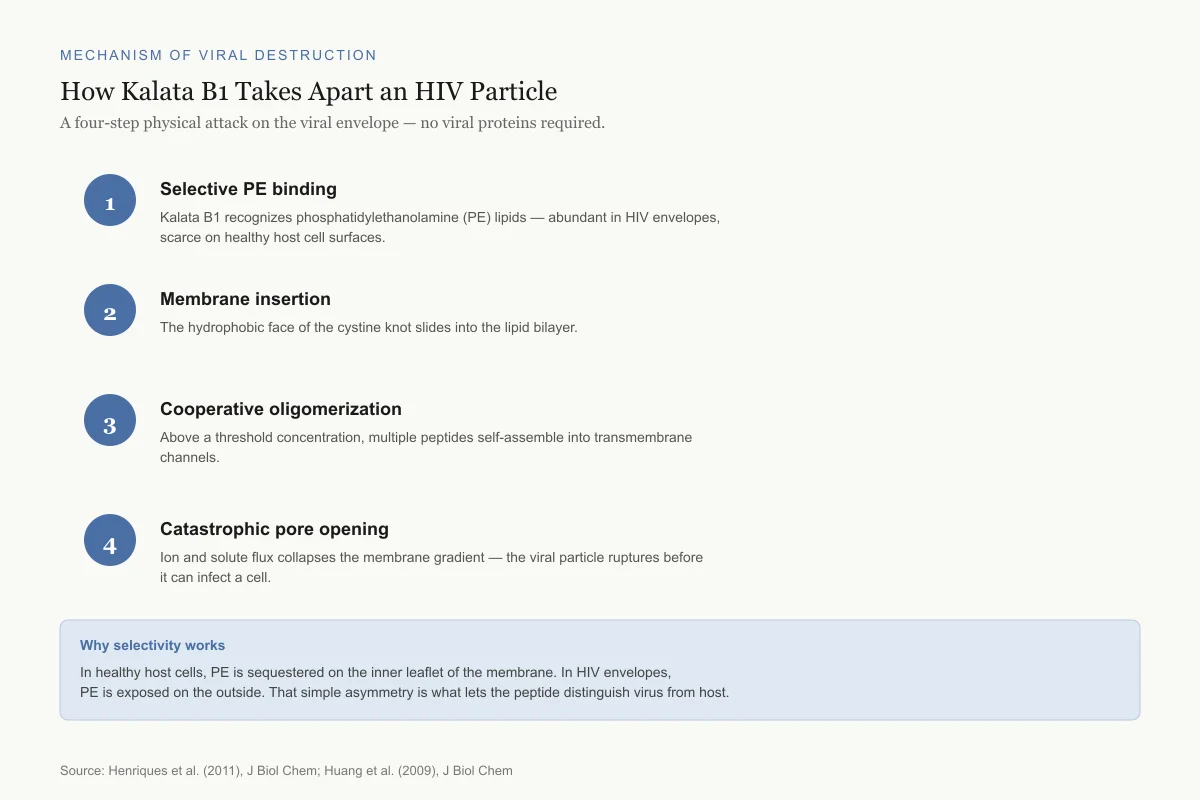
Mechanism of Viral Destruction
How Kalata B1 Takes Apart an HIV Particle
A four-step physical attack on the viral envelope — no viral proteins required.
Selective PE binding
Kalata B1 recognizes phosphatidylethanolamine (PE) lipids — abundant in HIV envelopes, scarce on healthy host cell surfaces.
Membrane insertion
The hydrophobic face of the cystine knot slides into the lipid bilayer.
Cooperative oligomerization
Above a threshold concentration, multiple peptides self-assemble into transmembrane channels.
Catastrophic pore opening
Ion and solute flux collapses the membrane gradient — the viral particle ruptures before it can infect a cell.
Why selectivity works
In healthy host cells, PE is sequestered on the inner leaflet of the membrane. In HIV envelopes, PE is exposed on the outside. That simple asymmetry is what lets the peptide distinguish virus from host.
Source: Henriques et al. (2011), J Biol Chem; Huang et al. (2009), J Biol Chem
 View as image
View as imageAnti-HIV activity: the evidence
The anti-HIV activity of cyclotides was first reported in the early 2000s and has been characterized most thoroughly for kalata B1 and cycloviolacin variants from Viola species.
Henriques et al. (2011) measured kalata B1's anti-HIV EC50 at approximately 2.5 micromolar against HIV-1 in cell-based assays. The antiviral effect was not mediated by interaction with viral proteins (gp120, gp41) or host cell receptors (CD4, CCR5, CXCR4). Instead, pre-incubation of HIV virions with kalata B1 prior to cell exposure abolished infectivity, confirming that the peptide acts directly on the viral particle rather than on the target cell.[2]
This mechanism distinguishes cyclotides from every class of approved antiretroviral drug. Existing HIV therapies target viral enzymes (reverse transcriptase, protease, integrase) or the interaction between viral surface proteins and host cell receptors (entry inhibitors). Cyclotides bypass these targets entirely by destroying the viral envelope before the virus can contact a cell. A virus with a ruptured membrane cannot infect anything, regardless of the state of its surface proteins or internal enzymes.
Mechanism Comparison
Cyclotides Attack a Different Part of the Virus
Every approved HIV drug targets a viral protein. Cyclotides target a physical property of the viral membrane.
Entry inhibitors
Target
gp120, gp41, CD4, CCR5
What it does
Block viral surface protein–host receptor binding
How resistance arises
Viral surface protein mutations
Reverse transcriptase inhibitors
Target
HIV reverse transcriptase enzyme
What it does
Stop conversion of viral RNA to DNA
How resistance arises
Enzyme active-site mutations
Integrase inhibitors
Target
HIV integrase
What it does
Prevent viral DNA from entering the host genome
How resistance arises
Integrase mutations
Protease inhibitors
Target
HIV protease
What it does
Block cleavage of viral polyproteins into functional parts
How resistance arises
Protease active-site mutations
Cyclotides (kalata B1)
Orthogonal mechanismTarget
PE-rich lipids on the viral envelope
What it does
Physical destruction of the viral membrane
How resistance arises
Would require full remodeling of viral envelope lipid composition
Because the target is a fundamental lipid composition rather than a specific protein, the evolutionary path to resistance is far steeper for cyclotides than for any existing drug class. A virus cannot mutate its way out of physics.
Source: Henriques et al. (2011); De Veer et al. (2019); standard antiretroviral drug class references
 View as image
View as imageSafety
ModerateNarrow gap between killing virus and damaging host cells
Concern
Cyclotides that puncture viral envelopes can also damage red blood cells and other host membranes at concentrations not far above the antiviral dose. Henriques et al. showed both the hemolytic and anti-HIV effects depend on the same phosphatidylethanolamine-binding mechanism.
What the research says
Structural engineering has partially separated these activities by tuning membrane binding vs. pore formation, but no cyclotide has yet achieved a therapeutic window suitable for human antiviral use.
Particularly relevant for: Any future human antiviral application — not currently a consumer exposure concern
What to do
This is a research-stage molecule. There are no approved or clinically tested cyclotide antivirals. Anyone encountering cyclotide products online should treat them as unapproved research chemicals.
Henriques et al. (2011), Journal of Biological Chemistry
De Veer et al. (2019) reviewed the broader antiviral evidence and noted that multiple cyclotides from different plant sources demonstrated anti-HIV activity, with cycloviolacins from Viola species often showing greater potency than the prototypic kalata B1. The structure-activity relationship data consistently linked antiviral potency to the surface hydrophobicity of the cyclotide and its affinity for PE-containing membranes.[1]
The key limitation is the therapeutic index. Cyclotides that disrupt viral membranes can also damage host cell membranes at higher concentrations, leading to hemolysis and cytotoxicity. The gap between antiviral EC50 and cytotoxic CC50 is narrow for some cyclotides, requiring careful optimization. Henriques et al. showed that the hemolytic and anti-HIV activities of kalata B1 are both PE-dependent but can be partially separated through structural modifications that alter the ratio of membrane binding to pore formation.[2]
Beyond HIV: host defense and insecticidal roles
Cyclotides are not niche antiviral molecules. They are broad-spectrum chemical weapons in plant defense.
Craik (2012) reviewed the host-defense activities of cyclotides in Toxins, documenting their roles against insects, nematodes, fungi, and bacteria. The best-characterized defense function is insecticidal activity. When Helicoverpa armigera larvae (a major agricultural pest) were fed artificial diets containing kalata B1 at micromolar concentrations, larval growth was reduced by over 50% and mortality increased. The mechanism was consistent with membrane disruption in the insect midgut, where epithelial cells were visibly damaged.[3]
The agricultural implications are substantial. Conventional chemical pesticides face resistance evolution, environmental persistence, and toxicity concerns. Cyclotides are biodegradable peptides produced by the plants themselves, and their CCK structure provides the stability needed to survive in the plant tissue and the insect gut. Craik (2010) discussed the dual potential of cyclotides in drug design and agriculture, arguing that their natural insecticidal function could be harnessed for crop protection through transgenic expression of cyclotide genes in crop plants.[6]
Antibacterial activity adds a third dimension. Multiple cyclotides exhibit antimicrobial effects against gram-positive and gram-negative bacteria at concentrations comparable to their antiviral activity, following the same PE-dependent membrane disruption mechanism. The fact that cyclotides target a fundamental physical property of microbial membranes (lipid composition) rather than a specific protein receptor makes resistance evolution through target mutation substantially more difficult. A bacterium would need to remodel its entire membrane lipid composition to evade cyclotide attack, a far higher evolutionary barrier than mutating a single enzyme or receptor.
Nematicidal activity has also been documented. Root-knot nematodes, which cause billions of dollars in crop losses annually, are susceptible to cyclotides at low micromolar concentrations. The nematode cuticle and intestinal membranes appear to be vulnerable to the same pore-forming mechanism that operates against insect midgut cells and viral envelopes.[3]
Mollica et al. (2015) characterized cyclotides as a "natural combinatorial peptide library," noting that the conserved CCK scaffold tolerates substantial sequence variation in the loops between cysteine residues. Nature has already performed a massive combinatorial experiment across hundreds of plant species, generating cyclotides with diverse loop sequences and correspondingly diverse biological activities while maintaining the structural core that confers stability and membrane affinity.[7]
The cyclotide scaffold for drug design
The exceptional stability of cyclotides has attracted drug designers seeking to solve one of the central problems in peptide therapeutics: rapid degradation in the body. Most therapeutic peptides are destroyed within minutes by proteases in the blood and gastrointestinal tract, limiting them to injectable administration. Cyclotides survive these environments.
Craik (2012) laid out the case for cyclotides as drug scaffolds in Expert Opinion on Drug Discovery. The approach, called "grafting," inserts a pharmacologically active peptide sequence into one of the exposed loops of the cyclotide scaffold, replacing the natural sequence while retaining the CCK core. The grafted sequence gains the stability of the cyclotide framework: resistance to proteases, thermal stability, and potentially oral bioavailability. The cyclotide scaffold acts as a molecular armored vehicle for the grafted peptide cargo.[13]
De Veer et al. (2017) expanded on this concept in Accounts of Chemical Research, describing cyclotides as "tools in chemical biology" and documenting successful grafting of bradykinin receptor agonists, melanocortin receptor ligands, and protease-resistant versions of bioactive peptide sequences into cyclotide scaffolds. In each case, the grafted cyclotide retained both the target activity of the inserted sequence and the stability properties of the scaffold.[10]
Muratspahic et al. (2020) applied this approach to G protein-coupled receptor (GPCR) ligands, demonstrating that cyclotide-grafted peptides could activate GPCRs with nanomolar potency while resisting proteolytic degradation that would inactivate the linear peptide equivalents. GPCRs are the target of approximately 35% of all approved drugs, making a protease-resistant peptide delivery platform for GPCR ligands particularly valuable.[8]
Koehbach et al. (2024) advanced the grafting methodology with a "plug and play" approach to chemical synthesis of grafted cyclotides, enabling rapid construction of cyclotide variants with different grafted sequences. This modular synthesis strategy reduces the time and cost of generating cyclotide drug candidates, potentially accelerating the transition from scaffold concept to clinical candidate.[12]
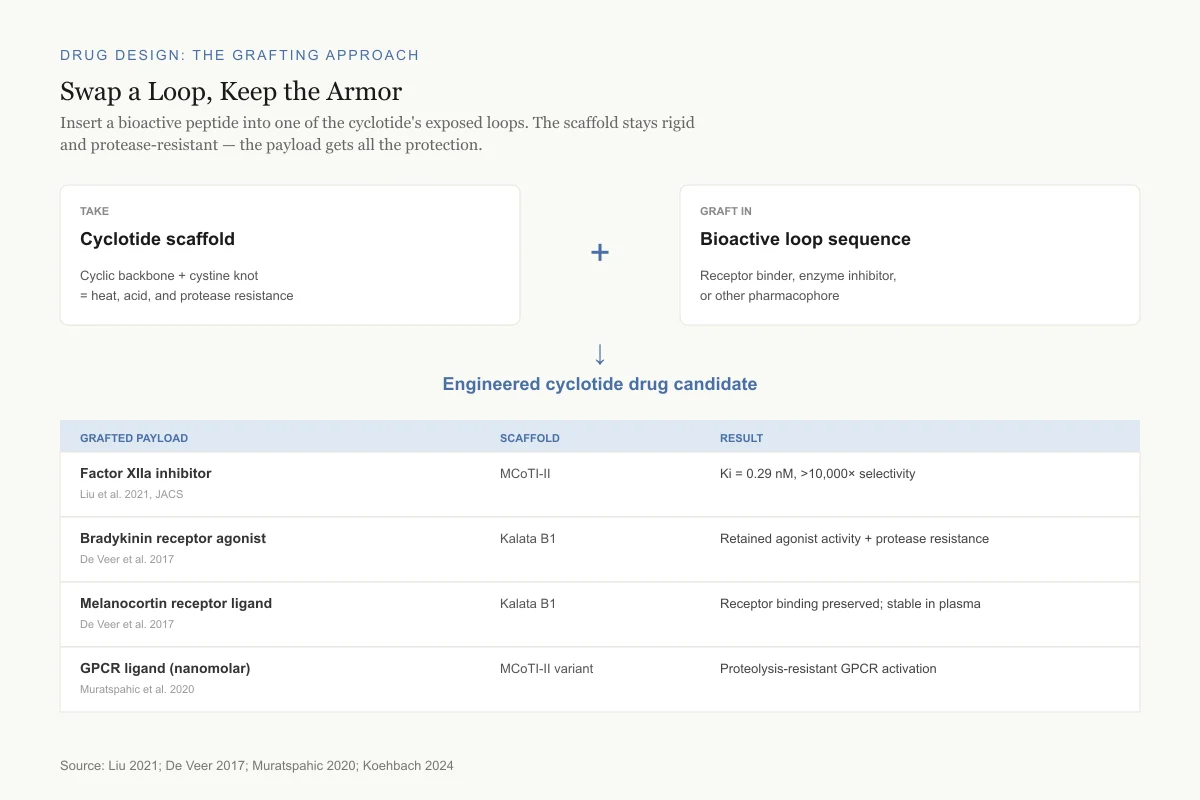
Drug Design: The Grafting Approach
Swap a Loop, Keep the Armor
Insert a bioactive peptide into one of the cyclotide's exposed loops. The scaffold stays rigid and protease-resistant — the payload gets all the protection.
Take
Cyclotide scaffold
Cyclic backbone + cystine knot = heat, acid, and protease resistance
Graft in
Bioactive loop sequence
Receptor binder, enzyme inhibitor, or other pharmacophore
Engineered cyclotide drug candidate
Grafted payload
Scaffold
Result
Factor XIIa inhibitor
Liu et al. 2021, JACS
MCoTI-II
Ki = 0.29 nM, >10,000× selectivity
Bradykinin receptor agonist
De Veer et al. 2017
Kalata B1
Retained agonist activity + protease resistance
Melanocortin receptor ligand
De Veer et al. 2017
Kalata B1
Receptor binding preserved; stable in plasma
GPCR ligand (nanomolar)
Muratspahic et al. 2020
MCoTI-II variant
Proteolysis-resistant GPCR activation
Source: Liu 2021; De Veer 2017; Muratspahic 2020; Koehbach 2024
 View as image
View as imageOral bioavailability: the cyclotide advantage
Most peptide drugs cannot be taken as pills because they are destroyed in the stomach and intestine. Cyclotides, evolved to survive in plant tissue and insect guts, resist this degradation.
Hyun et al. (2025) reviewed the evidence for cyclotides as scaffolds for orally active peptide therapeutics in Molecules and Cells. Grafted cyclotides have demonstrated oral bioactivity in animal models, with the CCK scaffold protecting the grafted sequence from gastrointestinal proteolysis. The review documented cases where cyclotide-grafted peptides administered orally produced pharmacological effects comparable to those achieved by injected linear peptides at similar doses.[9]
This oral stability is not trivial. The pharmaceutical industry has spent billions trying to develop oral peptide formulations through enteric coatings, protease inhibitor co-administration, permeation enhancers, and nanoparticle encapsulation. The cyclotide scaffold achieves protease resistance through molecular architecture alone, without requiring any of these formulation technologies. The cyclic backbone eliminates the free termini that exopeptidases attack, and the cystine knot protects the internal backbone from endopeptidase cleavage.
The remaining challenge is absorption. Surviving the GI tract is necessary but not sufficient for oral bioavailability; the peptide must also cross the intestinal epithelium into the bloodstream. Cyclotides are larger (approximately 3 kDa) than typical orally absorbed drugs, and their membrane-active properties, while useful for antiviral activity, must be carefully tuned to promote transcellular absorption without causing intestinal toxicity.
Engineered cyclotides as therapeutic candidates
The transition from natural cyclotide characterization to engineered therapeutics has accelerated.
Liu et al. (2021) published a striking example in the Journal of the American Chemical Society. They engineered an ultrapotent and selective inhibitor of human beta-Factor XIIa (a target in thrombosis) by grafting an optimized binding sequence into the MCoTI-II cyclotide scaffold. The resulting molecule inhibited Factor XIIa with picomolar potency (Ki = 0.29 nM) while showing over 10,000-fold selectivity against related serine proteases. The cyclotide framework provided both the structural rigidity needed for high-affinity binding and the protease resistance needed for potential in vivo use as an anticoagulant.[11]
This result illustrates the power of combining natural scaffold stability with rational sequence design. The Factor XIIa inhibitor would be rapidly degraded as a linear peptide. Embedded in the cyclotide scaffold, it retains picomolar potency while gaining the stability profile of a small protein. Amphibian skin peptides and other natural bioactive peptides face the same stability challenge; cyclotide scaffolding offers a general solution.
Koehbach et al. (2024) described a modular chemical synthesis approach that makes it practical to generate libraries of grafted cyclotides, each carrying a different bioactive sequence. This "plug and play" methodology allows researchers to rapidly screen cyclotide-delivered peptides against therapeutic targets, combining the stability of the natural scaffold with the diversity of combinatorial peptide design.[12]
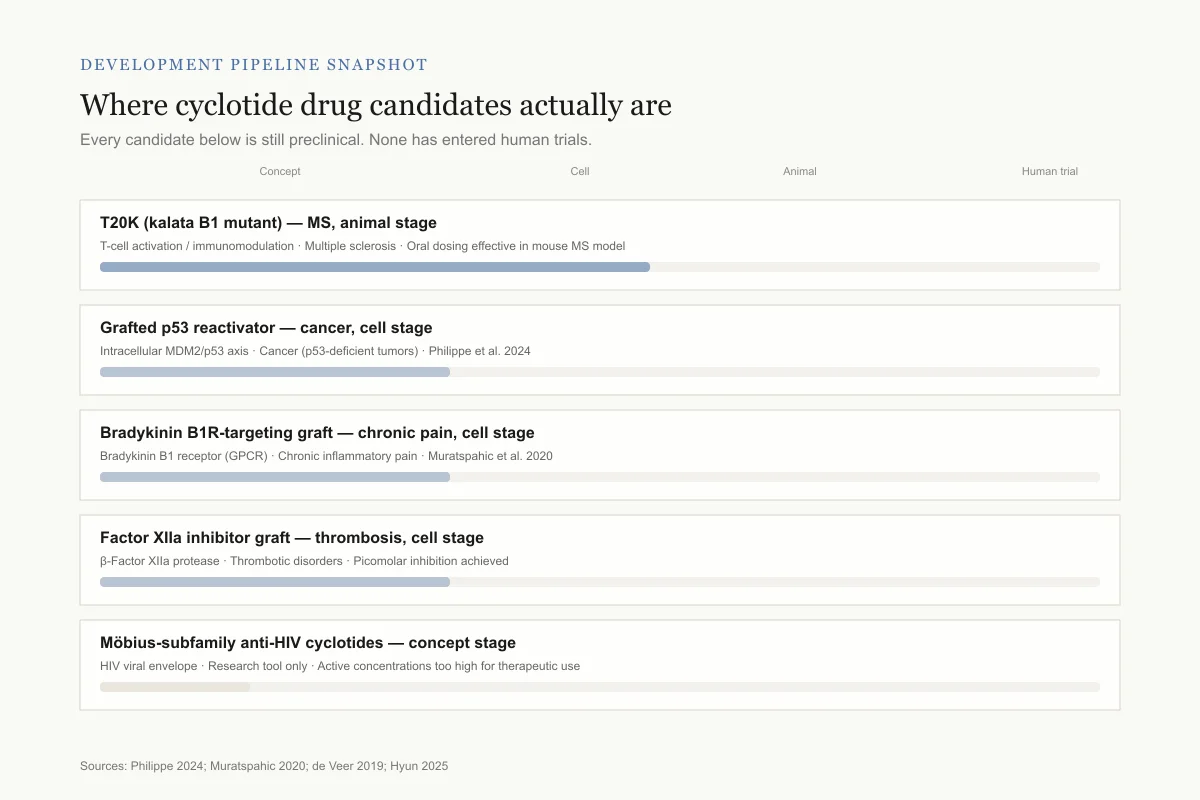
Development pipeline snapshot
Where cyclotide drug candidates actually are
Every candidate below is still preclinical. No cyclotide has entered human trials.
T20K (kalata B1 mutant)
Animal studiesTarget: T-cell activation / immunomodulation · Indication: Multiple sclerosis
Oral dosing effective in mouse MS model
Grafted cyclotide — p53 reactivator
Cell studiesTarget: Intracellular MDM2/p53 axis · Indication: Cancer (p53-deficient tumors)
Philippe et al. 2024
Bradykinin B1R-targeting graft
Cell studiesTarget: Bradykinin B1 receptor (GPCR) · Indication: Chronic inflammatory pain
Muratspahic et al. 2020
Factor XIIa inhibitor graft
Cell studiesTarget: Human β-Factor XIIa protease · Indication: Thrombotic disorders
Picomolar inhibition achieved
Native Möbius-subfamily cyclotides
ConceptTarget: HIV viral envelope · Indication: Anti-HIV (research tool only)
Active concentrations too high for therapeutic use
Biggest unknown: manufacturing. Making a correctly folded, triple-disulfide cyclic peptide at pharmaceutical scale is technically difficult and expensive — which is part of why no candidate has broken through to the clinic yet.
Sources: Philippe 2024; Muratspahic 2020; de Veer 2019; Hyun 2025
 View as image
View as imageWhat remains unknown
Despite two decades of intensive study, several fundamental questions about cyclotides remain open.
Selectivity optimization for antiviral use. The therapeutic index between antiviral activity and host cell toxicity is too narrow for most natural cyclotides to serve as drugs. Engineering cyclotide variants with improved selectivity for viral membranes over host cell membranes is an active but unsolved challenge. The PE-dependent selectivity described by Henriques et al. provides a starting point, but achieving clinically relevant selectivity ratios will require more precise control over membrane binding specificity than current structure-activity models allow.
In vivo antiviral efficacy. Most anti-HIV data for cyclotides come from cell-based assays. Published in vivo antiviral studies in animal models are limited. Whether the membrane-disrupting mechanism that works in a test tube translates to antiviral efficacy in the complex environment of blood plasma, where proteins compete for membrane binding and cyclotide concentrations may be diluted below the cooperative pore-formation threshold, is not established.
Immunogenicity. Cyclotides are foreign proteins. Whether repeated administration triggers immune responses that neutralize their activity or cause allergic reactions has not been systematically studied. Their small size (approximately 30 residues) may reduce immunogenicity compared to larger protein therapeutics, but this assumption requires validation.
Spectrum of antiviral activity. While anti-HIV activity is well documented, systematic evaluation against other enveloped viruses (influenza, SARS-CoV-2, dengue, Zika) is sparse. The PE-targeting mechanism should theoretically work against any virus with a PE-rich envelope, but the composition of viral membranes varies across virus families, and efficacy may differ substantially.
Manufacturing scale. Chemical synthesis of cyclotides is feasible but expensive due to the three disulfide bonds and head-to-tail cyclization. Recombinant production in plants or bacteria is under development but not yet optimized for pharmaceutical manufacturing scale. The grafting approach adds an additional synthetic step. Cost-effective manufacturing remains a barrier to clinical development.
Synergy with other antivirals. Cyclotides destroy viral particles through a mechanism entirely orthogonal to protease inhibitors, reverse transcriptase inhibitors, and entry inhibitors. Whether combining cyclotides with conventional antivirals produces synergistic effects has barely been explored. A membrane-disrupting agent that reduces viral load before particles reach target cells could theoretically lower the dose requirements for conventional drugs, reducing side effects while maintaining efficacy. This combination strategy warrants systematic investigation.
The Bottom Line
Cyclotides are ultra-stable plant peptides defined by a cyclic backbone and three interlocking disulfide bonds. Their antiviral activity operates through selective destruction of phosphatidylethanolamine-rich viral membranes, a mechanism demonstrated against HIV-1 at low micromolar concentrations. Beyond antiviral use, the cyclotide scaffold has been co-opted for drug design: grafted cyclotides can deliver bioactive peptide sequences with picomolar potency while surviving proteases and potentially enabling oral administration. The field has moved from structural characterization to engineered therapeutic candidates, though gaps remain in selectivity optimization, in vivo antiviral proof, and manufacturing scalability.
Sources & References
- 1RPEP-04145·de Veer, Simon J et al. (2019). “Cyclotides: From Structure to Function..” Chemical reviews.Study breakdown →PubMed →↩
- 2RPEP-01776·Henriques, Sónia Troeira et al. (2011). “Decoding the membrane activity of the cyclotide kalata B1: the importance of phosphatidylethanolamine phospholipids and lipid organization on hemolytic and anti-HIV activities..” The Journal of biological chemistry.Study breakdown →PubMed →↩
- 3RPEP-01923·Craik, David J (2012). “Host-defense activities of cyclotides..” Toxins.Study breakdown →PubMed →↩
- 4RPEP-00518·Craik, D J et al. (1999). “Discovery of Cyclotides: A New Family of Ultra-Stable Circular Peptides From Plants.” Journal of molecular biology.Study breakdown →PubMed →↩
- 5RPEP-01497·Huang, Yen-Hua et al. (2009). “The biological activity of the prototypic cyclotide kalata b1 is modulated by the formation of multimeric pores..” The Journal of biological chemistry.Study breakdown →PubMed →↩
- 6RPEP-01600·Craik, David J et al. (2010). “Cyclotides: Circular Peptides From Plants With Drug and Agricultural Applications.” Cellular and molecular life sciences : CMLS.Study breakdown →PubMed →↩
- 7RPEP-02742·Mollica, Adriano et al. (2015). “Cyclotides: Nature's Indestructible Peptides and Their Drug Discovery Potential.” Journal of enzyme inhibition and medicinal chemistry.Study breakdown →PubMed →↩
- 8RPEP-05017·Muratspahić, Edin et al. (2020). “Cyclotide Scaffolds Enable Orally Active Peptide Drugs Targeting GPCRs.” RSC chemical biology.Study breakdown →PubMed →↩
- 9RPEP-11493·Hyun, Youbong (2025). “Cyclotides as novel plant-derived scaffolds for orally active cyclic peptide therapeutics..” Molecules and cells.Study breakdown →PubMed →↩
- 10RPEP-03263·de Veer, Simon J et al. (2017). “Cyclotides as Tools in Chemical Biology..” Accounts of chemical research.Study breakdown →PubMed →↩
- 11RPEP-05562·Liu, Wenyu et al. (2021). “Ultrapotent Cyclic Peptide in Cyclotide Scaffold Blocks Blood Clotting Factor XIIa.” Journal of the American Chemical Society.Study breakdown →PubMed →↩
- 12RPEP-08573·Koehbach, Johannes et al. (2024). “A New 'Plug and Play' Method for Building Cyclotide-Based Peptide Drugs.” RSC chemical biology.Study breakdown →PubMed →↩
- 13RPEP-01922·Craik, David J et al. (2012). “Cyclotides as a basis for drug design..” Expert opinion on drug discovery.Study breakdown →PubMed →↩