Bulevirtide: The Peptide That Blocks Hepatitis D
Peptide Antivirals for Hepatitis
45% response at 48 weeks
In the Phase 3 MYR301 trial, 45% of patients receiving 2 mg bulevirtide daily achieved the combined virologic and biochemical endpoint at 48 weeks, versus 2% of controls.
Wedemeyer et al., NEJM, 2023
Wedemeyer et al., NEJM, 2023
If you only read one thing
Bulevirtide is a peptide drug — 47 amino acids with a fatty acid tail — that blocks hepatitis B and D viruses from getting into liver cells. It plugs the doorway the viruses use, a receptor called NTCP. For hepatitis D, which was untreatable for decades, a daily injection brings nearly half of patients to a combined virus-and-liver-enzyme response by one year. Europe approved it in 2020. The FDA has not approved it in the US as of early 2026.
Hepatitis D is the most severe form of viral hepatitis in humans. It accelerates cirrhosis, increases hepatocellular carcinoma risk, and until 2020, had no approved treatment. An estimated 12 to 72 million people worldwide carry the hepatitis D virus (HDV), which can only infect individuals already harboring hepatitis B virus (HBV) because HDV hijacks the HBV surface antigen to build its own viral envelope. For decades, the only option was interferon-alpha, a therapy with harsh side effects and response rates below 30%. Bulevirtide, sold as Hepcludex, changed that. It is a 47-amino-acid synthetic lipopeptide that blocks viral entry into liver cells by binding the sodium taurocholate cotransporting polypeptide (NTCP), the receptor both viruses use to enter hepatocytes.[1] Conditionally approved in the EU in July 2020 and granted full approval in 2023, bulevirtide represents the first drug ever designed specifically to treat hepatitis D, and only the second peptide-based antiviral to reach the market after enfuvirtide. This article covers its discovery, mechanism, clinical evidence, safety profile, and the open questions that remain. For more specific subtopics, see the cluster articles on HCV protease inhibitor peptides and peptide-based approaches to hepatitis B cure research.
Key Takeaways
- In the Phase 3 MYR301 trial, bulevirtide at 2 mg daily produced a combined virologic and biochemical response in 45% of patients at 48 weeks, compared to 2% receiving no treatment (Wedemeyer et al., NEJM, 2023).[1]
- HDV RNA became undetectable in 12% of the 2 mg group and 20% of the 10 mg group at 48 weeks, with responses continuing to improve through 96 weeks of treatment.[5]
- Real-world data from 114 patients showed a 76% virologic response rate, with a mean time to response of 23 weeks (Dietz-Fricke et al., JHEP Reports, 2023).[4]
- Cryo-EM imaging revealed that bulevirtide forms a plug-and-string structure on the NTCP receptor, with the myristoyl group anchoring into the lipid membrane and the peptide string covering the extracellular surface (Liu et al., Nature Communications, 2024).[3]
- An integrated safety analysis of 218 patients found no serious drug-related adverse events, with elevated bile acids and injection site reactions as the most common side effects (Asselah et al., Liver International, 2025).[6]
- No virologic resistance to bulevirtide was detected through 24 weeks of treatment in Phase II and III clinical trials (Hollnberger et al., J Hepatology, 2023).[12]
What is bulevirtide and why does it matter?
Bulevirtide (formerly known as Myrcludex B) is a synthetic lipopeptide consisting of 47 amino acids derived from the pre-S1 domain of the HBV large surface protein, with a myristoyl (C14 fatty acid) group attached to its N-terminus.[2] This structural design mimics the exact region of the HBV envelope that the virus uses to latch onto liver cells, effectively creating a molecular decoy that occupies the receptor before the virus can.
The drug emerged from Stephan Urban's laboratory at Heidelberg University, where researchers spent over a decade dissecting how HBV enters hepatocytes. Schulze et al. (2010) performed fine mapping of the pre-S1 sequence and identified that a conserved motif within the N-terminal 48 amino acids, combined with N-terminal myristoylation, was essential for both viral infectivity and the ability of synthetic peptides to block infection.[9] This work provided the structural blueprint for what would become bulevirtide.
The drug matters because hepatitis D has been an orphan disease in the truest sense. HDV is a defective virus, a satellite pathogen that encodes only two proteins and borrows HBV's surface antigen to form its own infectious particles. This dependency on HBV means HDV infection occurs exclusively in people already infected with HBV, either through simultaneous co-infection or superinfection. Despite affecting a relatively small fraction of the world's 296 million chronic HBV carriers, HDV causes the fastest-progressing form of viral hepatitis: up to 70% of HDV-positive patients develop cirrhosis within 5 to 10 years of infection.[11]
The NTCP discovery that made bulevirtide possible
The entire bulevirtide story depends on a single molecular discovery: the identification of NTCP as the receptor for HBV and HDV.
Yan et al. (2012) published the breakthrough in eLife. Using near-zero-distance photo-cross-linking and tandem affinity purification, the team identified NTCP (sodium taurocholate cotransporting polypeptide, also called SLC10A1) as the functional receptor for both HBV and HDV. When they silenced NTCP expression in susceptible cell lines, viral infection was abolished. When they forced NTCP expression in normally resistant hepatocarcinoma cells, those cells became susceptible to HBV and HDV infection.[7]
This was the missing piece. For decades, researchers knew that HBV required a liver-specific receptor but could not identify it because the virus's extreme species specificity (it infects only humans and a few primate species) made traditional receptor-hunting approaches difficult.
Ni et al. (2014) independently confirmed and extended the finding. Their work in Gastroenterology demonstrated that NTCP mediates species-specific entry for both HBV and HDV, identified two short sequence motifs in human NTCP required for viral binding, and showed that synthetic lipopeptides derived from pre-S1 could block the interaction.[8] This study established NTCP not just as the receptor, but as a druggable target.
NTCP's normal biological function is transporting bile salts from the blood into hepatocytes. It is expressed almost exclusively on the basolateral membrane of liver cells, which explains HBV's strict liver tropism. The virus evolved to exploit a transporter that is abundant only where it needs to replicate. Li and Urban (2016) reviewed how these entry insights opened up the first real therapeutic approaches for both HBV and HDV, noting that NTCP-targeting strategies could potentially block not just new infection but also the intrahepatic spread of virus between liver cells.[13]
How bulevirtide blocks viral entry at the molecular level
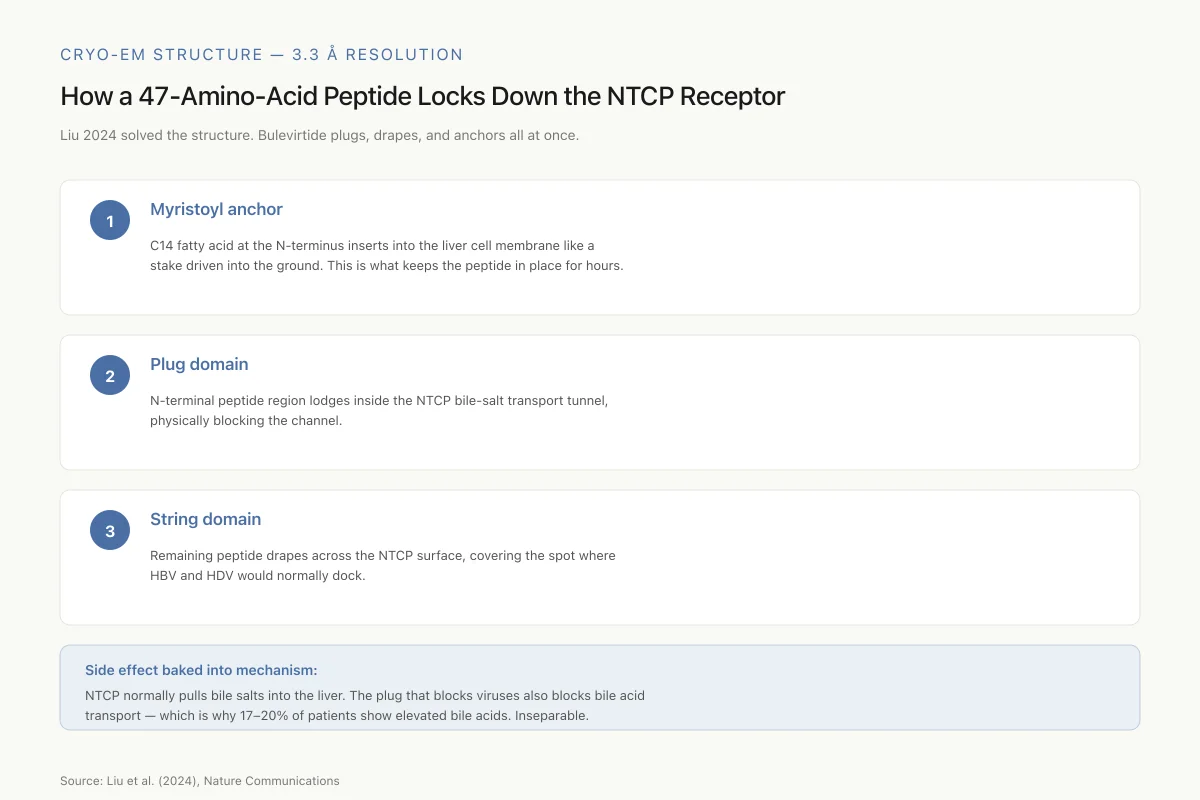
Cryo-EM Structure — 3.3 Å Resolution
How a 47-Amino-Acid Peptide Locks Down the NTCP Receptor
Liu 2024 solved the structure and found bulevirtide doesn’t just bind — it plugs, drapes, and anchors all at once.
C14 fatty acid at the N-terminus inserts into the liver cell membrane like a stake driven into the ground
N-terminal peptide region lodges inside the NTCP bile-salt transport tunnel, physically blocking the channel
Remaining peptide drapes across the NTCP surface, covering the spot where HBV and HDV would normally dock
Side effect baked into mechanism: NTCP normally pulls bile salts into the liver. The plug that blocks viruses also blocks bile acid transport — which is why 17–20% of patients show elevated serum bile acids. Inseparable.
Source: Liu et al. (2024), Nature Communications
 View as image
View as imageBulevirtide works by competitive inhibition. It binds to NTCP before the virus can, occupying the receptor and preventing HBV and HDV from docking and entering hepatocytes. But the molecular details, revealed only recently, show a more complex picture than simple receptor blocking.
Liu et al. (2024) solved the cryo-electron microscopy (cryo-EM) structure of bulevirtide bound to human NTCP at 3.3 angstrom resolution and published the results in Nature Communications. The structure revealed that bulevirtide forms two distinct domains when bound to the receptor. A "plug" domain, consisting of the peptide's N-terminal region, lodges directly into NTCP's bile salt transport tunnel, physically blocking the channel. A "string" domain, formed by the rest of the peptide, drapes across the receptor's extracellular surface, covering the area where the virus would normally bind.[3]
The N-terminal myristoyl group plays a critical structural role. Rather than pointing into the receptor, the fatty acid chain inserts into the lipid bilayer of the cell membrane adjacent to NTCP, anchoring the peptide in place like a stake driven into the ground. This dual anchoring, into both the receptor tunnel and the surrounding membrane, explains bulevirtide's tight binding and sustained occupancy of the receptor.[3]
This structural work also explains a known side effect. Because bulevirtide physically blocks NTCP's bile salt transport tunnel, it inhibits bile acid uptake into hepatocytes. This leads to elevated serum bile acid levels, the most common laboratory finding in clinical trials. The structural data confirm that antiviral activity and bile acid transport inhibition are mechanistically inseparable: any molecule that blocks viral entry through NTCP will also impair bile salt uptake, because the virus uses the transporter's own substrate channel.
The cryo-EM data also rationalized why certain naturally occurring NTCP variants in human populations are associated with reduced susceptibility to HBV infection. Mutations in the extracellular loops of NTCP that are covered by bulevirtide's string domain disrupt viral binding while partially preserving bile salt transport function.[3]
Phase 2 clinical evidence: the MYR202 trial
The first large randomized trial of bulevirtide was MYR202, a Phase 2 study published in The Lancet Infectious Diseases in 2023. Wedemeyer et al. enrolled 120 adults with chronic HDV across hospitals in Germany and Russia. Patients were randomized to receive 2 mg, 5 mg, or 10 mg subcutaneous bulevirtide daily in combination with tenofovir disoproxil fumarate (TDF), or TDF alone, for 24 weeks.[10]
The results demonstrated clear dose-dependent antiviral activity. At week 24, HDV RNA declined by at least 2 log10 from baseline in 54% of patients receiving 2 mg, 50% at 5 mg, 77% at 10 mg, and just 4% on TDF alone. ALT normalization, a marker of reduced liver inflammation, occurred in 43% to 50% of bulevirtide-treated patients.[10]
MYR202 established proof of concept for bulevirtide in humans and identified the 2 mg and 10 mg doses that would advance to Phase 3. The combination with TDF was designed to suppress HBV simultaneously, since HDV depends on HBV surface antigen for its lifecycle. However, the trial also showed that bulevirtide's anti-HDV effect did not translate into significant HBV surface antigen decline, suggesting that entry inhibition alone is insufficient to clear the HBV reservoir.
Phase 3 results: the MYR301 trial
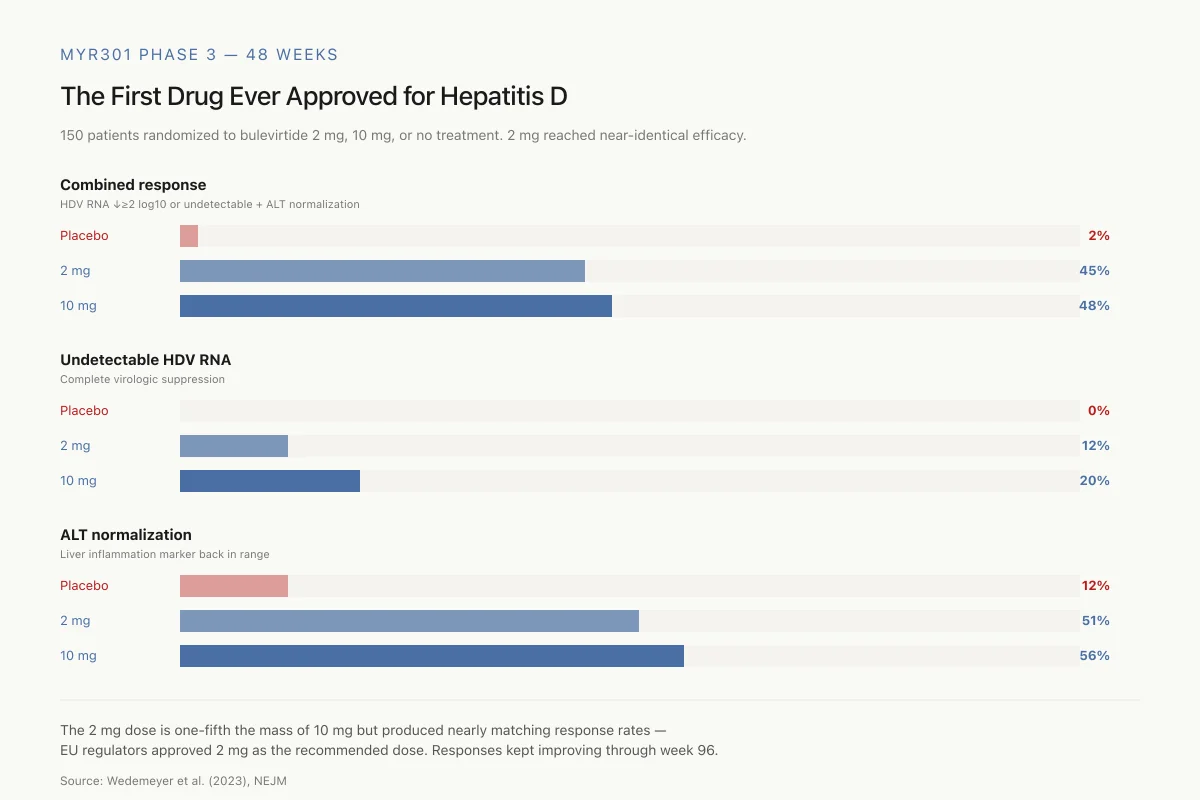
MYR301 Phase 3 — 48 Weeks
The First Drug Ever Approved for Hepatitis D
150 patients with chronic HDV randomized to bulevirtide 2 mg, 10 mg, or no treatment. The 2 mg dose reached nearly identical efficacy to 10 mg.
Combined response
HDV RNA ↓≥2 log10 or undetectable + ALT normalization
Undetectable HDV RNA
Complete virologic suppression
ALT normalization
Liver inflammation marker in range
The 2 mg dose is one-fifth the mass of 10 mg but produced nearly matching response rates — which is why EU regulators approved 2 mg as the recommended dose. Response rates continued improving through week 96.
Source: Wedemeyer et al. (2023), NEJM
 View as image
View as imageThe pivotal Phase 3 trial, MYR301, was published in the New England Journal of Medicine in 2023. This open-label study randomized 150 patients with chronic HDV (with or without compensated cirrhosis) to receive bulevirtide at 2 mg daily, 10 mg daily, or no treatment for 48 weeks followed by crossover to 10 mg for 96 weeks.[1]
The primary endpoint, a combined response of HDV RNA decline of at least 2 log10 or undetectable HDV RNA plus ALT normalization, was achieved by 45% of the 2 mg group and 48% of the 10 mg group at 48 weeks, compared to 2% in the control arm. For virologic response alone, HDV RNA became undetectable in 12% of the 2 mg group and 20% of the 10 mg group. ALT normalization occurred in 51% and 56%, respectively, versus 12% in controls.[1]
The MYR301 data carried several findings worth noting beyond the headline numbers. First, response rates continued to increase with treatment duration. The 48-week data represented an interim analysis of a planned 144-week treatment course, and longer follow-up showed further improvement. Second, even patients who did not achieve the combined endpoint often had meaningful virologic declines. Third, the 2 mg dose, which is one-fifth the mass of the 10 mg dose, produced nearly equivalent combined response rates, a finding that influenced the EU's decision to approve 2 mg as the recommended dose.
Extended follow-up: 96-week data
Wedemeyer et al. (2024) published the 96-week follow-up from MYR301 in the Journal of Hepatology. The extended data showed continued improvement across all response categories in both dose groups. Combined response rates rose from 45-48% at week 48 to higher levels at week 96, with particularly notable improvement in patients who had shown suboptimal virologic responses early in treatment.[5]
Liver stiffness, measured by transient elastography (FibroScan), also declined from week 48 through week 96, suggesting that sustained virologic suppression translates into measurable regression of liver fibrosis. This is a critical finding because the clinical value of any hepatitis treatment ultimately rests on its ability to prevent or reverse liver damage, not just suppress viral replication.[5]
The 96-week data also demonstrated that patients who began treatment late (the delayed-treatment control group that crossed over at week 48) achieved response rates comparable to those seen in the early-treatment groups at equivalent timepoints, confirming that the treatment effect is reproducible and not dependent on early initiation.
Real-world evidence beyond clinical trials
Clinical trials select patients carefully and monitor them intensively. Real-world data from routine clinical practice provides a different perspective.
Dietz-Fricke et al. (2023) reported outcomes from 114 patients treated with bulevirtide across German clinical centers. This cohort included patients who would have been excluded from the Phase 3 trial, including those with advanced liver disease and prior treatment failure. Despite this broader population, 76% achieved a virologic response (HDV RNA decline of at least 2 log10 or undetectable), with a mean time to response of 23 weeks.[4]
The real-world response rate of 76% exceeded the 45-48% combined endpoint in MYR301, partly because the real-world study used a less stringent endpoint (virologic response alone, without requiring simultaneous ALT normalization). Still, the data confirmed that bulevirtide's efficacy translates outside the controlled trial setting.
Of particular relevance: 90% of patients who achieved undetectable HDV RNA for 96 or more weeks while on treatment maintained that undetectable status after stopping bulevirtide. This durability of response raises the question of whether finite treatment courses, rather than lifelong therapy, might be sufficient for some patients.
Safety profile and tolerability
The tolerability of bulevirtide has been consistently favorable across trials. Asselah et al. (2025) published an integrated safety analysis in Liver International, pooling data from 218 patients across bulevirtide clinical trials at 48 weeks. No serious adverse events were attributed to the drug.[6]
The most common side effects were elevated total bile acid levels (reported in 17-20% of patients on bulevirtide versus 0% of controls) and injection site reactions (16-20%). Both are predictable consequences of the drug's mechanism. NTCP inhibition directly impairs hepatic bile acid uptake, leading to accumulation in the blood. And daily subcutaneous injections, regardless of the drug, commonly cause local reactions.[6]
The bile acid elevation warrants context. While elevated bile acids can cause pruritus (itching), the clinical significance of asymptomatic bile acid elevations during bulevirtide therapy is unclear. Bile acid levels returned to baseline after treatment discontinuation. No cases of bile acid-related liver toxicity were reported. However, the long-term consequences of chronically elevated bile acids during extended treatment courses (planned at 144 weeks or longer) remain an area to monitor.
Headache and pruritus were also reported more frequently in bulevirtide groups than controls, though the differences were modest. Overall discontinuation rates due to adverse events were low across all trials.
Safety
LowElevated bile acids are the cost of the mechanism
Concern
17–20% of patients on bulevirtide develop elevated serum bile acids. That’s not a random side effect — it’s how the drug works. NTCP also pumps bile salts into the liver, and the peptide blocks that job alongside blocking viral entry.
What the research says
Elevations have been clinically quiet in trials — no bile-acid-related liver toxicity reported through 96 weeks. Levels return to baseline after stopping. Pruritus is the main symptom if any.
Particularly relevant for: Anyone on bulevirtide long-term, especially if bile acid levels are being actively monitored
What to do
Expect routine bile acid monitoring. Report persistent itching. Long-term implications of chronic mild elevation remain under study.
Asselah et al. (2025); Liu et al. (2024) structural basis
No resistance detected through 24 weeks
One concern with any antiviral therapy is the emergence of drug-resistant viral variants. For bulevirtide, this concern is somewhat mitigated by its mechanism. The drug targets a host receptor (NTCP), not a viral protein. In theory, the virus cannot easily mutate its way around a host target.
Hollnberger et al. (2023) formally evaluated this by sequencing HDV RNA from patients in Phase II and III clinical trials through 24 weeks of treatment. No virologic resistance mutations were identified in any patient, including those with suboptimal virologic responses.[12]
This finding is consistent with the theoretical advantage of host-targeting antivirals over direct-acting antivirals. Whereas drugs targeting viral enzymes (like HCV protease inhibitors) face rapid resistance selection because the virus can mutate its own proteins, a drug that targets a human receptor faces a fundamentally different resistance landscape. HDV would need to evolve the ability to enter hepatocytes through an entirely different receptor, a much higher evolutionary barrier. Whether this resistance advantage holds over longer treatment durations remains to be confirmed.
Drug discovery and development timeline
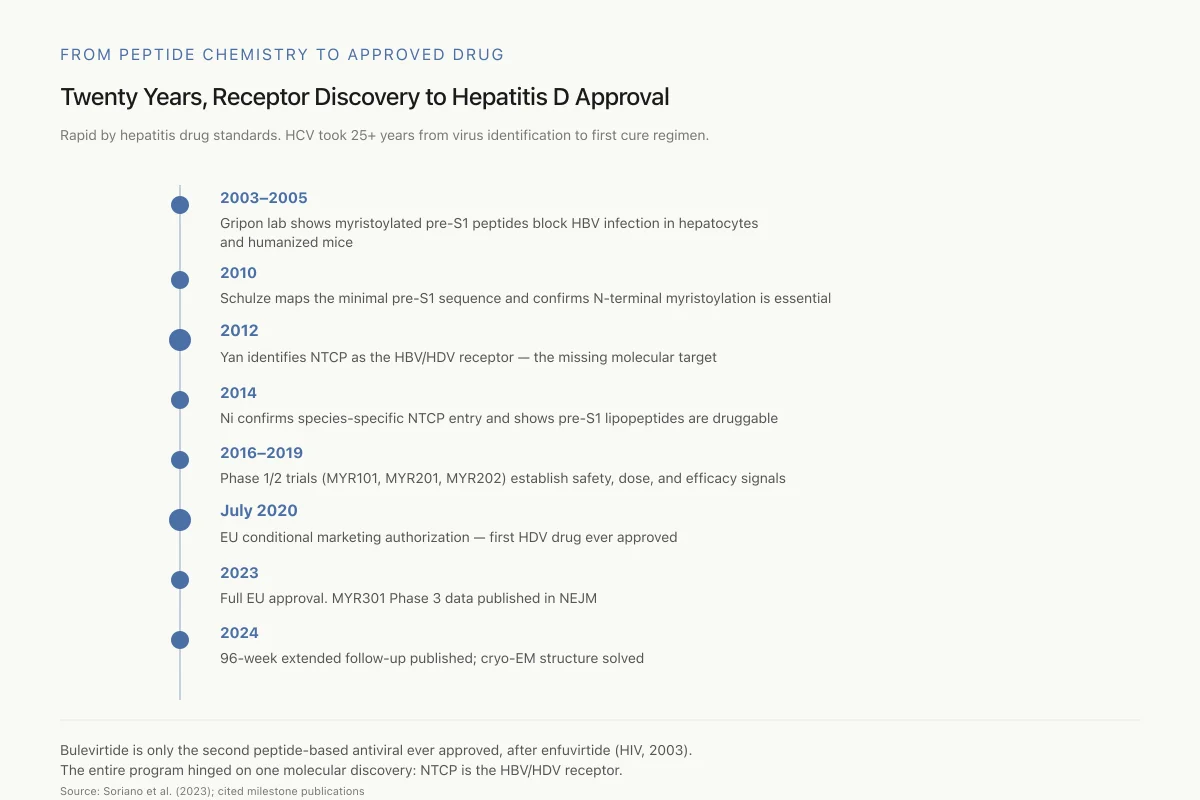
From Peptide Chemistry to Approved Drug
Twenty Years, Receptor Discovery to Hepatitis D Approval
Rapid by hepatitis drug standards. HCV took 25+ years from virus identification to its first curative regimen.
Bulevirtide is only the second peptide-based antiviral ever approved, after enfuvirtide (HIV, 2003). Its approval hinged on one molecular discovery — that NTCP is the HBV/HDV receptor — without which none of it happens.
Source: Soriano et al. (2023); cited milestone publications
 View as image
View as imageSoriano et al. (2023) published a comprehensive review of bulevirtide's path from laboratory to clinic in Drug Design, Development and Therapy. The timeline spans over two decades.[11]
Key milestones:
- 2003-2005: Gripon and colleagues at Heidelberg demonstrated that myristoylated pre-S1 peptides could block HBV infection in primary human hepatocytes and in humanized mouse models.
- 2010: Schulze et al. mapped the minimal pre-S1 sequence requirements and identified the critical role of N-terminal myristoylation.[9]
- 2012: Yan et al. identified NTCP as the receptor, providing the molecular target.[7]
- 2014: Ni et al. confirmed species-specific entry through NTCP and demonstrated druggability.[8]
- 2016-2019: Phase I and II clinical trials (MYR101, MYR201, MYR202) established safety, dose selection, and initial efficacy signals.
- 2020: EU conditional marketing authorization, making bulevirtide the first approved treatment for hepatitis D.
- 2023: Full EU approval based on Phase 3 MYR301 data. Publication of the NEJM trial results.
- 2024: 96-week extended follow-up data published. Cryo-EM structure solved.
The progression from basic peptide chemistry through receptor discovery to regulatory approval in under 20 years is rapid by hepatitis drug development standards. By comparison, effective HCV treatments took over 25 years from the virus's identification in 1989 to the first pan-genotypic cure regimens in 2014.
What bulevirtide does not do
Understanding bulevirtide's limitations is as important as understanding its efficacy.
Bulevirtide does not clear HBV surface antigen. Because the drug blocks new viral entry but does not affect HBV's covalently closed circular DNA (cccDNA) reservoir in already-infected hepatocytes, HBV surface antigen levels remain largely unchanged during treatment. This means bulevirtide alone cannot achieve functional cure of HBV.[1]
Bulevirtide does not eliminate HDV from all patients. Even at 96 weeks, a substantial proportion of patients still have detectable HDV RNA, although levels may be significantly reduced. Whether extended treatment beyond 144 weeks can achieve higher undetectability rates is under investigation.
Bulevirtide requires daily subcutaneous injection. Unlike oral antivirals such as the direct-acting agents used for HCV, bulevirtide must be self-administered via injection. For a chronic condition requiring extended treatment, this creates adherence challenges. Development of longer-acting formulations or alternative delivery routes has not been publicly announced.
Bulevirtide's effect on clinical outcomes (cirrhosis progression, liver cancer, mortality) has not been directly demonstrated. The fibrosis improvement signal from FibroScan measurements at 96 weeks is encouraging, but formal clinical outcome trials have not been completed. The drug's approval was based on surrogate virologic and biochemical endpoints.
Comparison with other peptide antivirals
Bulevirtide is only the second peptide-based antiviral to achieve regulatory approval, after enfuvirtide (Fuzeon), the HIV-1 fusion inhibitor approved in 2003. The comparison is instructive.
Both drugs are injectable peptides that block viral entry. Both target the host cell membrane interaction step rather than intracellular viral replication. Both were developed after the identification of their target mechanism through basic research. And both face the practical challenge of daily injection in conditions that require chronic treatment.
Enfuvirtide was eventually relegated to salvage therapy for multi-drug-resistant HIV because oral alternatives became available. Bulevirtide's trajectory may differ because HDV currently has no effective oral alternative, and the NTCP-targeting mechanism is not easily replicated by a small molecule (the receptor's bile acid transport function and viral binding involve complex protein-protein interactions that peptides are uniquely suited to disrupt).
The antiviral peptides against influenza field illustrates the broader potential of peptide-based viral entry inhibitors, though none have reached clinical approval for influenza. What distinguishes bulevirtide is that it crossed the translational gap from lab peptide to approved drug, a journey that most antiviral peptide candidates never complete.
Open questions and ongoing research
Several questions remain for bulevirtide and the NTCP-targeting approach:
Optimal treatment duration. The current recommendation is indefinite treatment, but the real-world data showing sustained response off-treatment in some patients suggests that finite treatment might be feasible. Biomarkers that predict which patients can safely stop treatment are not yet established.
Combination strategies. Bulevirtide combined with pegylated interferon-alpha has shown higher response rates than monotherapy in preliminary data. Whether combining entry inhibition with immune stimulation can achieve functional HDV cure is an active area of clinical investigation.
HBV functional cure. Because bulevirtide blocks NTCP and thus new infection cycles, it may play a role in combination strategies aimed at HBV functional cure (HBsAg loss). The drug alone does not achieve this, but it could prevent reinfection of hepatocytes while other agents target the intracellular HBV reservoir. For more on this topic, see peptide-based approaches to hepatitis B cure research.
Long-term safety of NTCP inhibition. NTCP is the primary hepatic bile acid transporter. Chronic inhibition raises bile acid levels and could theoretically affect cholesterol metabolism, fat-soluble vitamin absorption, or bile acid signaling pathways over years. The 96-week safety data are reassuring, but longer follow-up is needed.
Pediatric use. HDV infection in children, particularly in high-endemicity regions of Central Asia and sub-Saharan Africa, is underserved. No pediatric data for bulevirtide have been published.
The Bottom Line
Bulevirtide is the first peptide drug designed to block hepatitis virus entry at the NTCP receptor, and the first approved treatment of any kind for chronic hepatitis D. Phase 3 data show 45-48% combined response rates at 48 weeks with continued improvement through 96 weeks, and real-world evidence confirms efficacy in broader patient populations. The drug's safety profile is favorable, with no serious drug-related events and no detected resistance mutations. Structural data now reveal exactly how the lipopeptide blocks both viral entry and bile acid transport. Key unknowns include optimal treatment duration, the role of combination therapy in achieving functional cure, and the long-term metabolic consequences of sustained NTCP inhibition.
Sources & References
- 1RPEP-07537·Wedemeyer, Heiner et al. (2023). “A Phase 3, Randomized Trial of Bulevirtide in Chronic Hepatitis D..” The New England journal of medicine.Study breakdown →PubMed →↩
- 2RPEP-04893·Kang, Connie et al. (2020). “Bulevirtide: First Approval..” Drugs.Study breakdown →PubMed →↩
- 3RPEP-08739·Liu, Hongtao et al. (2024). “Structure of antiviral drug bulevirtide bound to hepatitis B and D virus receptor protein NTCP..” Nature communications.Study breakdown →PubMed →↩
- 4RPEP-06841·Dietz-Fricke, Christopher et al. (2023). “Treating hepatitis D with bulevirtide - Real-world experience from 114 patients..” JHEP reports : innovation in hepatology.Study breakdown →PubMed →↩
- 5RPEP-09513·Wedemeyer, Heiner et al. (2024). “Bulevirtide monotherapy in patients with chronic HDV: Efficacy and safety results through week 96 from a phase III randomized trial..” Journal of hepatology.Study breakdown →PubMed →↩
- 6RPEP-10004·Asselah, Tarik et al. (2025). “Bulevirtide Monotherapy Is Safe and Well Tolerated in Chronic Hepatitis Delta: An Integrated Safety Analysis of Bulevirtide Clinical Trials at Week 48..” Liver international : official journal of the International Association for the Study of the Liver.Study breakdown →PubMed →↩
- 7RPEP-02107·Yan, Huan et al. (2012). “Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus..” eLife.Study breakdown →PubMed →↩
- 8RPEP-02460·Ni, Yi et al. (2014). “Hepatitis B and D viruses exploit sodium taurocholate co-transporting polypeptide for species-specific entry into hepatocytes..” Gastroenterology.Study breakdown →PubMed →↩
- 9RPEP-01689·Schulze, Andreas et al. (2010). “Fine mapping of pre-S sequence requirements for hepatitis B virus large envelope protein-mediated receptor interaction..” Journal of virology.Study breakdown →PubMed →↩
- 10RPEP-07538·Wedemeyer, Heiner et al. (2023). “Bulevirtide: How a Peptide Entry Blocker Fights the World's Worst Hepatitis Virus.” The Lancet. Infectious diseases.Study breakdown →PubMed →↩
- 11RPEP-07407·Soriano, Vicente et al. (2023). “Bulevirtide in the Treatment of Hepatitis Delta: Drug Discovery, Clinical Development and Place in Therapy..” Drug design.Study breakdown →PubMed →↩
- 12RPEP-06954·Hollnberger, Julius et al. (2023). “Hepatitis Delta Virus Shows No Resistance to the Peptide Drug Bulevirtide After 24 Weeks.” Journal of hepatology.Study breakdown →PubMed →↩
- 13RPEP-03014·Li, Wenhui et al. (2016). “How Scientists Found the Doorway Hepatitis B Uses to Enter Liver Cells — And a Peptide to Block It.” Journal of hepatology.Study breakdown →PubMed →↩