ANP: The Atrial Peptide That Lowers Blood Pressure
Natriuretic Peptide Hormones
28 Amino Acids
Atrial natriuretic peptide is a 28-amino-acid hormone released by cardiac atria in response to volume overload. It reduces blood pressure through vasodilation, natriuresis, and aldosterone suppression, acting as a direct counter-regulatory system to the renin-angiotensin-aldosterone axis.
Pagel et al., J Cardiothorac Vasc Anesth, 2026
Pagel et al., J Cardiothorac Vasc Anesth, 2026
If you only read one thing
Your heart isn't just a pump — it's also a hormone-making gland. When the upper chambers stretch from too much blood volume, they release a peptide called ANP that tells blood vessels to relax, kidneys to dump salt and water, and the hormone system that raises blood pressure to back off. In heart failure, ANP is actually high, but the body's 'off switch' gets overwhelmed. That's why the leading heart failure drug (Entresto, a.k.a. sacubitril/valsartan) works by blocking the enzyme that destroys ANP — it gives the body's own pressure-lowering signal more time to work, and it cuts heart failure deaths and hospitalizations by about 20%.
In 1981, Adolfo de Bold injected atrial tissue extracts into rats and observed a rapid, profound drop in blood pressure accompanied by massive natriuresis. The substance responsible was a 28-amino-acid peptide now called atrial natriuretic peptide (ANP), and its discovery fundamentally changed how cardiovascular medicine understood blood pressure regulation. Before ANP, the heart was viewed primarily as a pump. After ANP, it became clear that the heart is also an endocrine organ that secretes peptide hormones to regulate its own workload.[1]
Four decades later, ANP's biology underpins one of the most consequential heart failure drugs of the past decade: sacubitril/valsartan (Entresto), which works by blocking the enzyme that degrades natriuretic peptides.[2] This article covers ANP's structure, signaling, physiological roles, and clinical relevance as the founding member of the natriuretic peptide family. For the clinical biomarker applications, see BNP and NT-proBNP: The Heart Failure Blood Tests Explained. For how the natriuretic peptide system protects organs beyond the heart, see How Natriuretic Peptides Protect Your Heart and Kidneys.
Key Takeaways
- Your heart isn't just a pump. It's also a hormone-making gland — a fact cardiovascular medicine only figured out in 1981.
- When the heart's upper chambers stretch from too much blood volume, they release a peptide called ANP that pushes blood pressure back down.
- ANP does three jobs at once: relaxes blood vessels, tells kidneys to dump salt and water, and shuts off the hormones that raise blood pressure.
- It's the body's natural counterweight to the blood-pressure-raising system. Without it, your pressure regulation would tip one way only.
- In heart failure, ANP levels are actually high — but the body's off-switch gets overwhelmed. This is what allows the disease to progress.
- The leading heart failure drug (Entresto) works by blocking the enzyme that destroys ANP, giving your own signal more time to work. It cuts deaths by 20%.
- Doctors also use related peptides (BNP, NT-proBNP) as a blood test for heart failure — used millions of times a year to guide diagnosis.
Structure and Synthesis
ANP is synthesized as a 151-amino-acid precursor called pre-proANP in atrial cardiomyocytes. Cleavage of the signal peptide produces proANP (126 amino acids), which is stored in dense secretory granules in the atrial wall. When the atria stretch due to increased blood volume, proANP is released and cleaved by the transmembrane serine protease corin into the biologically active 28-amino-acid C-terminal fragment (ANP) and an inactive 98-amino-acid N-terminal fragment (NT-proANP).[1]
The mature ANP peptide contains a 17-amino-acid disulfide-bonded ring structure (formed between cysteine residues at positions 7 and 23) that is essential for receptor binding. This ring structure is conserved across all natriuretic peptide family members and defines the pharmacophore that activates the guanylyl cyclase receptor.
ANP circulates with a half-life of approximately 2 to 3 minutes, making it one of the shortest-lived peptide hormones in human physiology. Clearance occurs through two mechanisms: enzymatic degradation by neprilysin (neutral endopeptidase, NEP) and receptor-mediated internalization through the natriuretic peptide clearance receptor (NPR-C). This extremely short half-life means that plasma ANP levels reflect real-time atrial stretch, making ANP a dynamic indicator of volume status rather than a cumulative biomarker.
The storage mechanism is distinctive among peptide hormones. Most peptide hormones are synthesized on demand or stored in small quantities. Atrial cardiomyocytes, by contrast, contain dense secretory granules packed with proANP, visible under electron microscopy as characteristic electron-dense structures near the nucleus. A single human atrium contains enough stored proANP to produce a measurable hemodynamic response within minutes of release. This storage-and-release model allows ANP to function as an emergency responder to acute volume overload, providing a rapid signal that does not depend on new gene transcription or protein synthesis.
The cleavage enzyme corin is itself regulated by cardiac conditions. In heart failure, corin expression is reduced and its activity diminished, which paradoxically impairs the processing of proANP to active ANP at precisely the moment when the compensatory response is most needed. Mutations in the corin gene have been associated with hypertension and preeclampsia in genetic studies, providing independent confirmation that the ANP processing pathway is critical for blood pressure homeostasis.
The Natriuretic Peptide Family
ANP was the first natriuretic peptide discovered, but two additional family members have since been identified.
Brain natriuretic peptide (BNP) is a 32-amino-acid peptide primarily synthesized in ventricular cardiomyocytes in response to ventricular wall stress. Despite its name (it was originally isolated from porcine brain tissue), BNP functions as a cardiac hormone with effects similar to ANP. BNP and its stable cleavage byproduct NT-proBNP are the natriuretic peptides used clinically as heart failure biomarkers. For their diagnostic and prognostic applications, see BNP and NT-proBNP: The Heart Failure Blood Tests Explained.
C-type natriuretic peptide (CNP) is a 22-amino-acid peptide expressed primarily in vascular endothelium rather than in the heart. Goetze and colleagues characterized CNP in a 2025 review as a paracrine/autocrine mediator rather than a classic blood-borne hormone, with distinct effects on vascular tone, endothelial function, and bone growth.[3] CNP signals through a different receptor (NPR-B) than ANP and BNP, giving it a distinct physiological profile. Kodal and colleagues designed a once-weekly CNP analog for the treatment of heart failure with preserved ejection fraction (HFpEF), demonstrating that the natriuretic peptide pharmacology extends beyond ANP and BNP.[4]
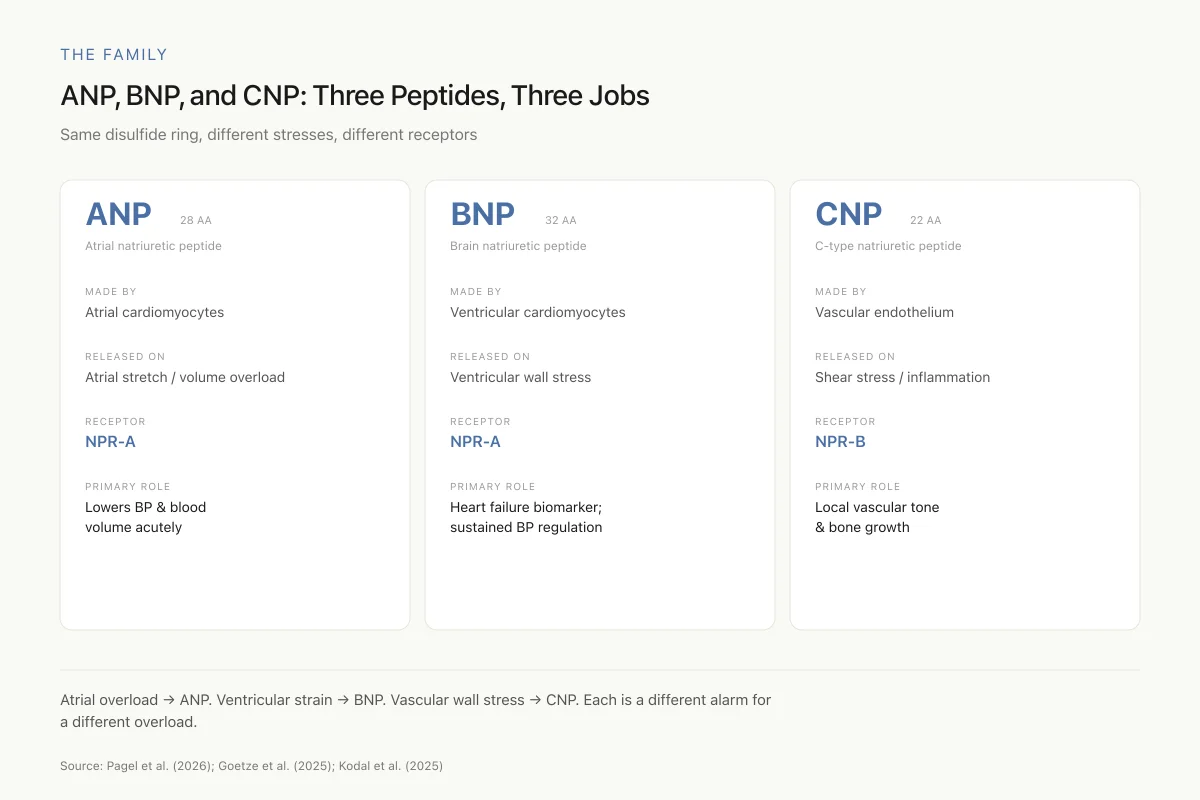
The Family
ANP, BNP, and CNP: Three Peptides, Three Jobs
All share the same disulfide ring structure, but they respond to different stresses and signal through different receptors
Think of the heart and blood vessels as having their own stress-response system: atrial overload → ANP, ventricular strain → BNP, vascular wall stress → CNP. Each peptide is a different alarm for a different overload.
Source: Pagel et al. (2026); Goetze et al. (2025); Kodal et al. (2025)
 View as image
View as imageThe three peptides form a complementary system: ANP responds primarily to atrial volume overload, BNP to ventricular wall stress, and CNP to vascular shear stress. Together, they provide the cardiovascular system with a peptide-based counter-regulatory mechanism against the vasoconstrictive, sodium-retaining effects of the renin-angiotensin-aldosterone system (RAAS).
A fourth natriuretic peptide, dendroaspis natriuretic peptide (DNP), was isolated from the venom of the green mamba snake in the 1990s. While not endogenous to mammals, DNP has been detected in human plasma and atrial tissue, though its physiological significance remains debated. DNP's extended C-terminal tail gives it unique pharmacological properties, and it has been explored as a template for drug design. Its existence illustrates the evolutionary conservation of natriuretic peptide signaling across vertebrates.
Understanding the natriuretic peptide family is essential for interpreting clinical biomarker data. When clinicians measure "BNP" or "NT-proBNP" in heart failure patients, they are measuring one member of a multi-peptide hormonal system. The clinical utility of these measurements depends on understanding that BNP elevation reflects ventricular wall stress, while ANP elevation reflects atrial pressure overload, and that both peptides are simultaneously active in the circulation. For a comprehensive discussion of these biomarker applications, see BNP and NT-proBNP: The Heart Failure Blood Tests Explained.
How ANP Lowers Blood Pressure
ANP activates the natriuretic peptide receptor A (NPR-A), a transmembrane guanylyl cyclase that generates cyclic GMP (cGMP) as its second messenger. The downstream effects of ANP-NPR-A-cGMP signaling reduce blood pressure through four parallel mechanisms.
Vasodilation. ANP relaxes vascular smooth muscle through cGMP-mediated reduction of intracellular calcium, causing direct vasodilation of both arteries and veins. The venous dilation is particularly important because it reduces cardiac preload (venous return), directly decreasing the volume of blood the heart must pump.
Natriuresis and diuresis. ANP acts on the kidney to increase sodium and water excretion. It dilates the afferent arteriole while constricting the efferent arteriole of the glomerulus, increasing glomerular filtration rate. It simultaneously inhibits sodium reabsorption in the collecting duct. The net effect is a reduction in blood volume. Inoue and colleagues demonstrated in 2025 that deletion of natriuretic peptide/guanylyl cyclase-A signaling in mice worsened diabetic kidney disease, confirming that the ANP signaling pathway has direct renoprotective effects beyond its blood pressure-lowering action.[5]
Aldosterone suppression. ANP directly inhibits aldosterone secretion from the adrenal cortex. Because aldosterone promotes sodium retention and potassium excretion, suppressing it amplifies the natriuretic effect and reduces the RAAS-driven sodium retention that characterizes heart failure and hypertension.
Renin inhibition. ANP suppresses renin release from the juxtaglomerular cells of the kidney, reducing the upstream activation of the entire RAAS cascade. By acting at the top of the RAAS hierarchy, this single inhibitory signal cascades downstream to reduce angiotensin I, angiotensin II, and aldosterone production simultaneously. This makes ANP a functional antagonist of the RAAS at multiple levels.
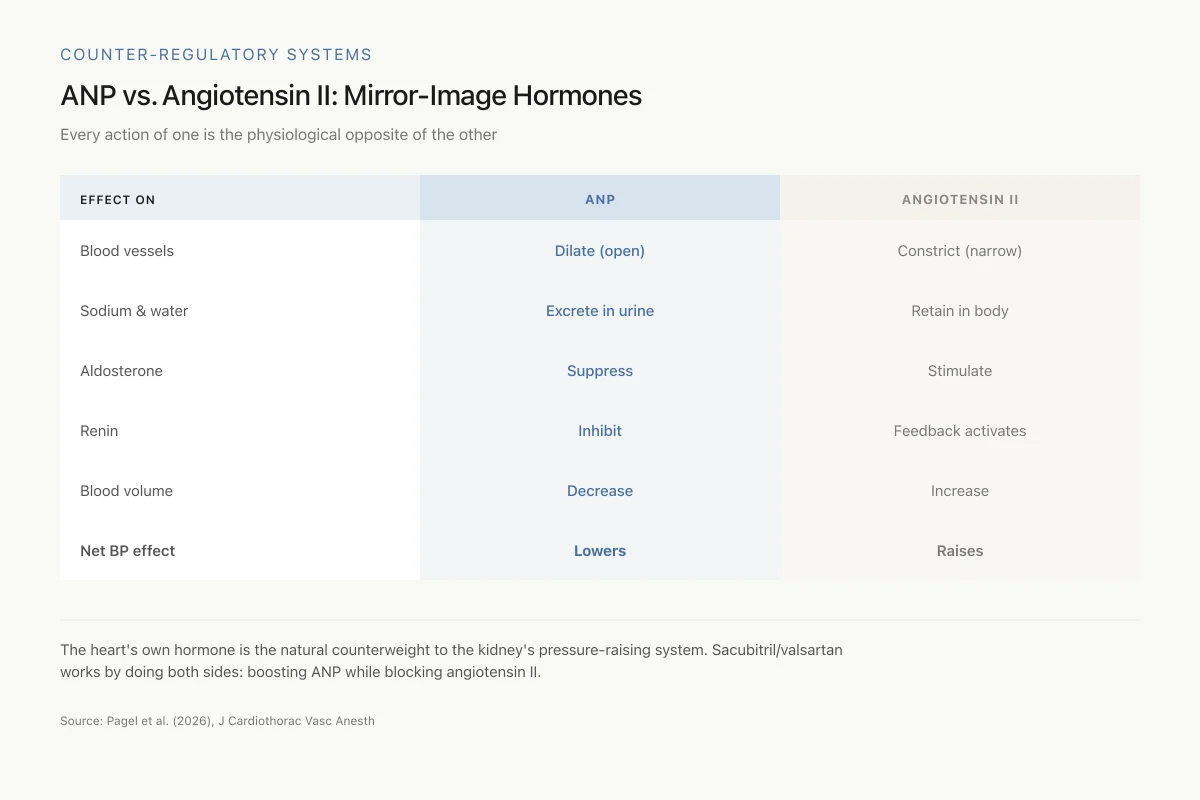
Counter-Regulatory Systems
ANP vs. Angiotensin II: Mirror-Image Hormones
Every action of one is the physiological opposite of the other
The heart's own hormone (ANP) is the natural counterweight to the kidney's pressure-raising system (renin-angiotensin-aldosterone). Sacubitril/valsartan works by doing both sides of this equation at once: boosting ANP (via neprilysin inhibition) while blocking angiotensin II.
Source: Pagel et al. (2026), J Cardiothorac Vasc Anesth
 View as image
View as imageThese four mechanisms position ANP as the physiological opposite of angiotensin II. Where angiotensin II constricts vessels, retains sodium, stimulates aldosterone, and increases blood volume, ANP dilates vessels, excretes sodium, suppresses aldosterone, and reduces blood volume. The balance between these two systems determines hemodynamic homeostasis.
Beyond these classical hemodynamic effects, ANP exerts direct effects on cardiac structure. It inhibits cardiac fibroblast proliferation and collagen synthesis, reducing pathological cardiac remodeling. It suppresses cardiomyocyte hypertrophy through cGMP-dependent inhibition of calcineurin/NFAT signaling, one of the primary pathways driving maladaptive ventricular growth. These anti-fibrotic and anti-hypertrophic effects are independent of blood pressure reduction and contribute to the structural cardiac benefits observed with therapies that enhance natriuretic peptide signaling.
ANP also has anti-inflammatory properties. It reduces endothelial expression of adhesion molecules, decreases leukocyte adhesion to activated endothelium, and modulates macrophage polarization. These effects have implications for atherosclerosis, where inflammatory cell recruitment to the vascular wall drives plaque formation. The anti-inflammatory profile of ANP suggests that natriuretic peptides participate in vascular protection beyond their hemodynamic role.
ANP in Disease: Heart Failure and Beyond
In heart failure, ANP levels are elevated, not deficient. The failing heart stretches under volume and pressure overload, triggering increased ANP and BNP secretion as a compensatory response. However, this compensatory mechanism becomes insufficient because the RAAS is simultaneously activated to a greater degree, because neprilysin degrades the natriuretic peptides before they can fully exert their effects, and because receptor desensitization occurs with chronic elevation.
Knany and colleagues examined a less recognized effect of ANP in heart failure: its impact on the lungs. Their 2025 study documented that ANP attenuates alveolar fluid clearance by reducing epithelial sodium channel activity in lung tissue, a finding with implications for understanding pulmonary edema in heart failure patients.[6] This paradoxical effect, where a peptide that clears fluid from the vascular space may impair fluid clearance from the lungs, illustrates the complexity of natriuretic peptide biology in disease states.
The elevated ANP levels in heart failure are not just a marker of disease severity. They represent an active but overwhelmed compensatory response. Studies have shown that the degree of ANP elevation correlates with prognosis: patients with higher ANP levels at diagnosis have worse outcomes, not because ANP is harmful but because higher levels indicate greater cardiac stress. The therapeutic strategy of enhancing natriuretic peptide activity (through neprilysin inhibition) works precisely because the endogenous compensatory response is insufficient on its own, and amplifying it provides additional hemodynamic relief.
ANP and metabolic regulation
Beyond cardiovascular effects, ANP participates in metabolic regulation. Natriuretic peptides promote lipolysis in adipose tissue through cGMP-dependent activation of hormone-sensitive lipase. This lipid-mobilizing effect is independent of sympathetic nervous system activation and represents a direct hormonal link between cardiac function and energy metabolism. Epidemiological observations that lower natriuretic peptide levels are associated with higher body mass index, insulin resistance, and incident diabetes have led to the concept of a "natriuretic peptide deficiency state" in obesity that may contribute to cardiometabolic risk.
This metabolic dimension connects ANP biology to the broader peptide hormone landscape. Just as GLP-1 receptor agonists were initially developed for diabetes and subsequently found cardiovascular benefit, ANP's metabolic effects suggest that natriuretic peptide enhancement may have metabolic benefits beyond its primary cardiovascular indications.
The ANP/BNP ratio as a clinical marker
Yamada and colleagues introduced the ANP-to-BNP ratio as a marker of atrial remodeling severity in 2026. In patients with atrial fibrillation and left atrial enlargement, the ANP/BNP ratio stratified atrial remodeling severity and predicted post-ablation recurrence risk. Because ANP is primarily atrial in origin while BNP is primarily ventricular, the ratio provides information about the relative contribution of atrial versus ventricular dysfunction that neither peptide alone can capture.[7]
Badoz and colleagues similarly evaluated MR-proANP (a stable fragment of the ANP precursor) as a predictor of sinus rhythm maintenance after electrical cardioversion for atrial fibrillation. Their 2025 multicenter study found that MR-proANP levels before cardioversion predicted whether patients would maintain sinus rhythm at one year, supporting the clinical utility of ANP-derived markers in arrhythmia management.[8]
Sacubitril/Valsartan: Pharmacology Built on ANP Biology
The most clinically impactful application of ANP biology is sacubitril/valsartan (Entresto), approved for heart failure in 2015. Sacubitril inhibits neprilysin, the enzyme that degrades ANP, BNP, and CNP. By blocking neprilysin, sacubitril increases circulating natriuretic peptide levels, amplifying their vasodilatory, natriuretic, and anti-fibrotic effects. Valsartan blocks the angiotensin II receptor, suppressing the RAAS. The combination simultaneously enhances the protective natriuretic peptide system while suppressing the harmful RAAS.[1]
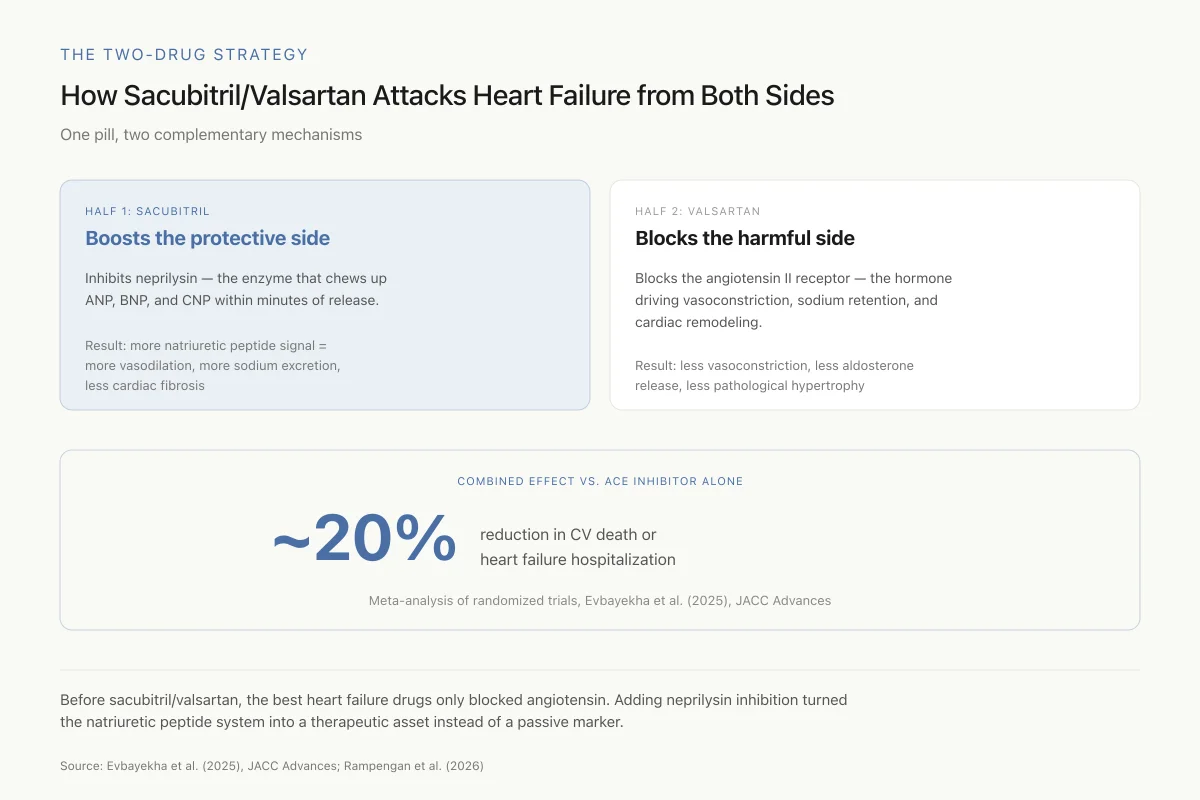
The Two-Drug Strategy
How Sacubitril/Valsartan Attacks Heart Failure from Both Sides
One pill, two complementary mechanisms — the first successful attempt to boost natriuretic peptides and block angiotensin simultaneously
Before sacubitril/valsartan, the best heart failure drugs blocked angiotensin II or its production. Adding neprilysin inhibition turned the natriuretic peptide system into a therapeutic asset instead of a passive marker — proving the whole concept of targeting this pathway.
Source: Evbayekha et al. (2025), JACC Advances; Rampengan et al. (2026)
 View as image
View as imageEvbayekha and colleagues published a systematic review and meta-analysis in JACC Advances in 2025, pooling data from randomized trials comparing sacubitril/valsartan against ACE inhibitors or ARBs in heart failure. The analysis confirmed superiority of sacubitril/valsartan for reducing the composite of cardiovascular death or heart failure hospitalization, with a relative risk reduction of approximately 20%.[2]
Rampengan and colleagues extended this analysis in 2026, evaluating the cardiorenal safety and efficacy of ARNIs across heart failure phenotypes. Their review demonstrated that sacubitril/valsartan's benefits span the ejection fraction spectrum, from HFrEF (reduced ejection fraction) to HFpEF (preserved ejection fraction), though the magnitude of benefit varies by phenotype. The renal protective effects were consistent across subgroups, reflecting the direct role of natriuretic peptides in maintaining glomerular filtration and tubular sodium handling.[9]
Safety
ModerateNeprilysin inhibition is not selective
Concern
Sacubitril doesn't only boost natriuretic peptides — it also raises bradykinin (angioedema risk), substance P (cough), and amyloid-beta (theoretical long-term cognitive concern). The drug must be started at least 36 hours after stopping an ACE inhibitor to avoid overlapping angioedema risk.
What the research says
The cardiovascular benefit is real and well-documented. The side effect profile is manageable but genuine — angioedema is rare but serious. Long-term Alzheimer's risk is theoretical, and no signal has emerged in post-marketing surveillance through ~2026.
Particularly relevant for: Patients starting or switching to sacubitril/valsartan, especially those with prior ACE-inhibitor angioedema
What to do
Never combine with ACE inhibitors. Wait at least 36 hours after stopping an ACE inhibitor before starting sacubitril/valsartan. Report swelling of face, lips, or throat immediately.
Sacubitril/valsartan prescribing information; Rampengan et al. (2026)
The success of sacubitril/valsartan has validated the concept of natriuretic peptide enhancement as a therapeutic strategy, but it has also revealed limitations. Neprilysin inhibition is non-selective: it increases levels of all neprilysin substrates, not just natriuretic peptides. The consequence is that bradykinin levels rise (contributing to the risk of angioedema), amyloid-beta clearance may be affected (raising theoretical concerns about long-term cognitive effects), and substance P levels increase (contributing to cough in some patients). These off-target effects reflect the fundamental challenge of targeting a degradation enzyme rather than the receptor pathway directly.
Beyond sacubitril: the future of natriuretic peptide pharmacology
The success of neprilysin inhibition has prompted exploration of alternative strategies to enhance natriuretic peptide signaling. These include direct NPR-A agonists (small molecules that activate the guanylyl cyclase receptor without requiring the native peptide), soluble guanylyl cyclase stimulators (which enhance cGMP production downstream of the receptor), and phosphodiesterase-9 inhibitors (which prevent cGMP degradation, amplifying the signal from whatever natriuretic peptide is present). Each approach has a different selectivity profile and a different set of potential off-target effects.
The long-acting CNP analog developed by Kodal and colleagues represents another direction: using a different member of the natriuretic peptide family as the therapeutic agent.[4] Because CNP signals through NPR-B rather than NPR-A, and because it is primarily an endothelial and skeletal peptide rather than a cardiac one, a CNP analog could provide some of the vascular benefits of natriuretic peptide enhancement without the cardiac volume effects of ANP/BNP. Whether this receptor selectivity translates to a different clinical profile is the central question of the ongoing development program.
Neprilysin beyond the heart
The neprilysin enzyme does not exclusively degrade natriuretic peptides. It also cleaves bradykinin, substance P, endothelin, amyloid-beta, and various other peptide substrates. Wu and colleagues reviewed neprilysin's role in musculoskeletal diseases in 2026, demonstrating that the enzyme regulates inflammation and tissue remodeling in contexts far removed from cardiovascular medicine.[10] This broad substrate profile explains both the efficacy and the side effects of neprilysin inhibition: by blocking one enzyme, sacubitril affects multiple peptide systems simultaneously.
Gao and colleagues demonstrated a specific protective mechanism of BNP in 2026, showing that brain natriuretic peptide protects against pulmonary vasoconstriction during acute pulmonary embolism through the natriuretic peptide receptor-A/cGMP pathway. Using both in vivo and ex vivo pulmonary artery models, they provided direct evidence that natriuretic peptides actively oppose the vasoconstrictive response to pulmonary emboli. The protection was mediated through cGMP-dependent smooth muscle relaxation in the pulmonary vasculature, a mechanism distinct from the systemic vasodilation that characterizes ANP's blood pressure-lowering effect. This finding has potential implications for acute PE management, where pulmonary artery resistance is the primary driver of right heart failure and death.[11]
What Remains Unknown
Despite four decades of research, several aspects of ANP biology remain unresolved. The mechanisms of ANP receptor desensitization in chronic heart failure are incompletely understood: why does the heart secrete more ANP while the peripheral response to ANP diminishes? Identifying the molecular basis of this resistance could reveal new therapeutic targets.
The relationship between natriuretic peptide deficiency and hypertension is established in animal models but incompletely characterized in humans. Can and colleagues' 2025 study demonstrated that surgical manipulation of the right atrial appendage (the primary site of ANP storage) directly affects circulating ANP and BNP levels, confirming that the atrial appendage is functionally important for natriuretic peptide homeostasis.[12] Whether variations in atrial appendage anatomy contribute to individual differences in natriuretic peptide levels and hypertension susceptibility has not been studied.
The interaction between natriuretic peptides and the emerging incretin-based therapies (GLP-1 receptor agonists, GIP/GLP-1 dual agonists) is another area of active investigation. Both natriuretic peptides and GLP-1 agonists reduce blood pressure, improve cardiac function, and promote weight loss, but through fundamentally different receptor systems and signaling pathways. Whether combining neprilysin inhibition with GLP-1 agonism would produce additive cardiovascular benefit or redundant effects has not been tested in a dedicated clinical trial, though observational data from patients receiving both therapies is accumulating.
The genetics of natriuretic peptide biology also present unresolved questions. Common genetic variants near the NPPA gene (encoding ANP) and the NPPB gene (encoding BNP) are associated with blood pressure variation in genome-wide association studies. Variants that increase natriuretic peptide levels are associated with lower blood pressure and reduced heart failure risk, while variants that decrease levels are associated with higher risk. These genetic associations suggest that individual differences in natriuretic peptide production may contribute meaningfully to cardiovascular risk stratification, potentially identifying patients who would benefit most from natriuretic peptide-enhancing therapies.
The therapeutic potential of direct natriuretic peptide replacement (as opposed to preventing degradation with neprilysin inhibitors) remains an open question. Nesiritide, a recombinant BNP, was approved for acute heart failure but showed no mortality benefit in the ASCEND-HF trial and has largely fallen out of use. For that story, see Nesiritide: The Recombinant BNP That Was Used to Treat Heart Failure. The development of long-acting CNP analogs by Kodal and colleagues suggests that the field has not abandoned direct natriuretic peptide agonism but is instead pursuing it through different members of the family and with improved pharmacokinetic profiles.[4]
For how natriuretic peptide biomarkers guide clinical decisions in real time, see Natriuretic Peptide-Guided Therapy: Using a Biomarker to Adjust Treatment.
The Bottom Line
ANP is a 28-amino-acid peptide hormone released by atrial cardiomyocytes that lowers blood pressure through vasodilation, natriuresis, aldosterone suppression, and renin inhibition. Its biology underpins sacubitril/valsartan therapy, which has demonstrated a 20% reduction in cardiovascular death or heart failure hospitalization. The natriuretic peptide system, including ANP, BNP, and CNP, continues to generate new therapeutic approaches beyond neprilysin inhibition.
Sources & References
- 1RPEP-15843·Pagel, Paul S et al. (2026). “Advances in Cardiovascular Pharmacotherapy. VI. Sacubitril-Valsartan, An Angiotensin Receptor-Neprilysin Inhibitor..” Journal of cardiothoracic and vascular anesthesia.Study breakdown →PubMed →↩
- 2RPEP-10872·Evbayekha, Endurance et al. (2025). “Sacubitril/Valsartan vs Standard Heart Drugs: Who Benefits Most from the Natriuretic Peptide Approach.” JACC. Advances.Study breakdown →PubMed →↩
- 3RPEP-11130·Goetze, Jens P et al. (2025). “Perspectives on C-Type Natriuretic Peptide in Cardiometabolic Disease..” Journal of the American Heart Association.Study breakdown →PubMed →↩
- 4RPEP-11881·Kodal, Anne Louise Bank et al. (2025). “A Once-Weekly C-Type Natriuretic Peptide for Treatment of Heart Failure with Preserved Ejection Fraction..” Journal of medicinal chemistry.Study breakdown →PubMed →↩
- 5RPEP-11525·Inoue, Yui et al. (2025). “Finerenone ameliorates diabetic kidney disease exacerbated by deletion of natriuretic peptide/guanylyl cyclase-A signaling and dietary high-protein load..” Scientific reports.Study breakdown →PubMed →↩
- 6RPEP-11870·Knany, Yara et al. (2025). “How Natriuretic Peptides Affect Fluid Clearance in the Lungs During Heart Failure.” International journal of molecular sciences.Study breakdown →PubMed →↩
- 7RPEP-16464·Yamada, Shinya et al. (2026). “Atrial to brain natriuretic peptide ratio as a marker of atrial remodeling severity and post-ablation recurrence risk in atrial fibrillation patients with left atrial enlargement..” Journal of interventional cardiac electrophysiology : an international journal of arrhythmias and pacing.Study breakdown →PubMed →↩
- 8RPEP-10047·Badoz, Marc et al. (2025). “Blood Peptide MR-proANP Predicts Whether Heart Rhythm Will Stay Normal After Cardioversion.” BMC cardiovascular disorders.Study breakdown →PubMed →↩
- 9RPEP-15969·Rampengan, Derren D C H et al. (2026). “Cardiorenal safety and efficacy of angiotensin receptor-neprilysin inhibitors in heart failure across the ejection fraction spectrum: A meta-analysis and meta-regression of RCTs with 28,001 patients..” Biomedical reports.Study breakdown →PubMed →↩
- 10RPEP-16436·Wu, Zuping et al. (2026). “Neprilysin: The Peptide-Degrading Enzyme That Affects Bones, Cartilage, and Muscles.” Tissue & cell.Study breakdown →PubMed →↩
- 11RPEP-15193·Gao, Yizhuo et al. (2026). “Brain Natriuretic Peptide Shows Therapeutic Potential for Acute Pulmonary Embolism.” Basic research in cardiology.Study breakdown →PubMed →↩
- 12RPEP-10288·Can, Murat Fatih et al. (2025). “Impact of Right Atrial Appendage Ligation vs. Repair on Serum Atrial Natriuretic Peptide, Brain Natriuretic Peptide, and Atrial Fibrillation following Coronary Artery Bypass Grafting..” Brazilian journal of cardiovascular surgery.Study breakdown →PubMed →↩